Epigenetics is defined as the study of inheritable changes in the gene expressions and phenotypes that occurs without altering the normal DNA sequence. These changes are mainly due to an alteration in chromatin or its packaging, which changes the DNA accessibility. DNA methylation, histone modification, and noncoding or microRNAs can best explain the mechanism of epigenetics. There are various DNA methylated enzymes, histone-modifying enzymes, and microRNAs involved in the cause of various CVDs (cardiovascular diseases) such as cardiac hypertrophy, heart failure, and hypertension. Moreover, various CVD risk factors such as diabetes mellitus, hypoxia, aging, dyslipidemia, and their epigenetics are also discussed together with CVDs such as CHD (coronary heart disease) and PAH (pulmonary arterial hypertension). Furthermore, different techniques involved in epigenetic chromatin mapping are explained.
1. Introduction
Epigenetics is defined as the study of inheritable changes in the gene expressions and phenotypes that occurs without altering the normal DNA sequence. These changes are mainly due to an alteration in chromatin or its packaging, which changes the DNA accessibility
[1]. These epigenetic changes are often due to the interactions of the genes with their surrounding conditions or the environment, causing either an increase or a decrease in gene expression or potentially leading to gene silencing as in the case of obesity, diabetes, and hypertension
[2].
Cardiovascular disease (CVD) epigenetics is considered to be a relatively new field. One of the leading causes of death worldwide, i.e., heart failure (HF), occurs when the myocardium undergoes functional and structural modifications. These processes result in the transcriptional and genomic reprogramming of cardiomyocytes and other neighboring cells
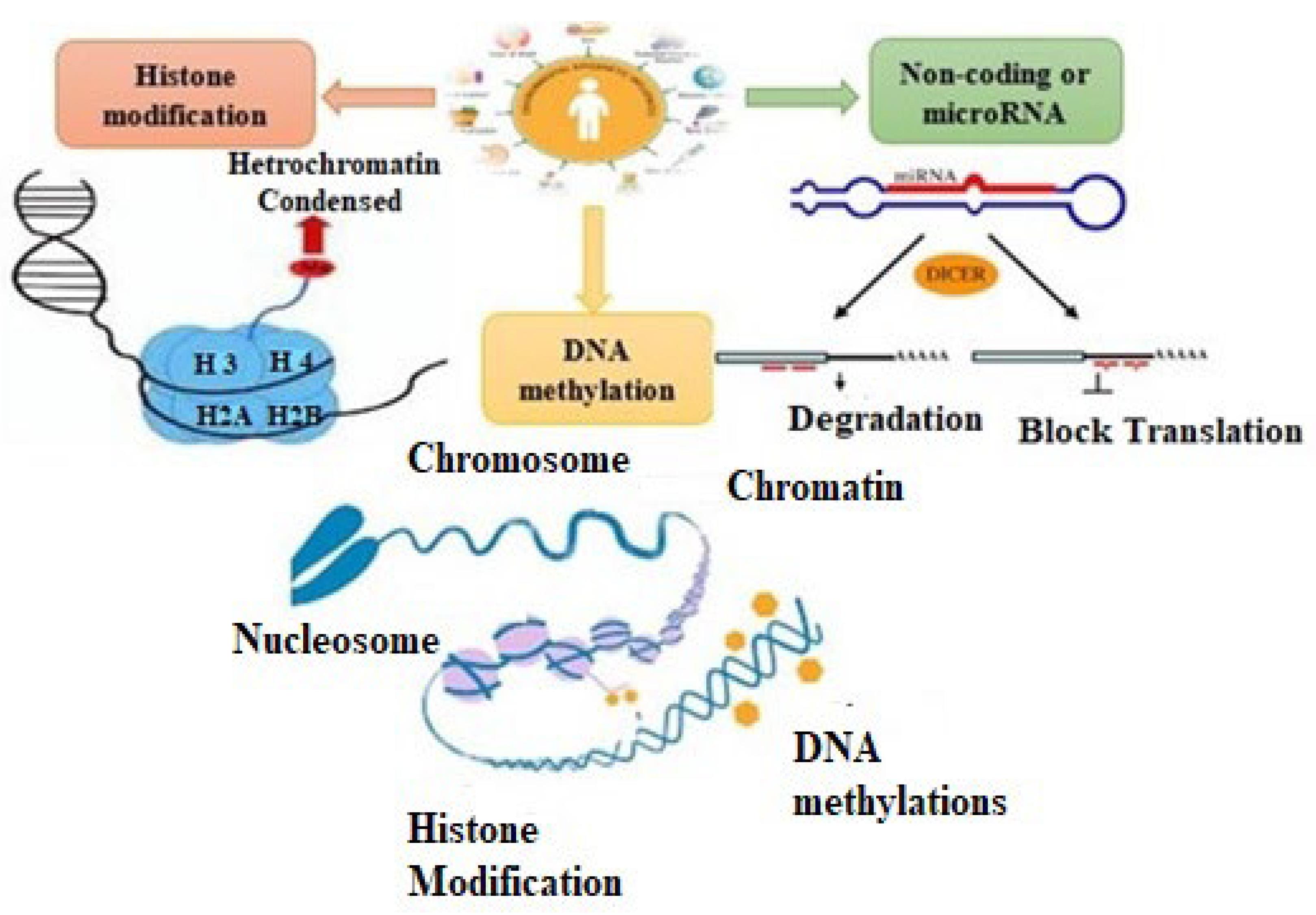
[3]. Due to a lack of knowledge in comprehending complex CVD pathophysiology, scientists are searching for other pathways. Epigenetic modifications of the genome represent one such pathway. The mechanisms of epigenetics can be best explained via (
Figure 1) DNA methylation, histone modification, and noncoding or microRNAs. They regulate gene expressions and affect the related risk factors, i.e., diabetes, hypertension, inflammation, and atherosclerosis. Unlike other genetic aberrations and mutations, epigenetic modifications are dynamic and can be altered either by therapeutic approaches or by lifestyle
[4].
Figure 1. Epigenetics and environmental factors.
1.1. DNA Methylation
DNA methylation basically occurs at cystine residues in CpG sequences at the fifth position. These CpG sequences, instead of localizing in the coding region, localize in the promoter region
[5]. This results in suppression of gene transcription either indirectly by recognizing the methylated sites using chromatin-modifying enzymes or directly by impeding the attachment of transcriptional factors to the promoter region in DNA
[6]. DNA methylation is mediated by DNA methyltransferases such as DNMT3b, DNMT3a, and DNMT1. Methylation status during replication is maintained by DNMT1, which recognizes the hypermethylated DNA. On the other hand, DNMT3a and DNMT3b are involved in de novo methylation
[7].
1.2. Histone Modification
Post-translational histone modification consists of phosphorylation, ubiquitination, acetylation, and methylation. These modifications occur in different patterns, regulating the shifting of the open chromatin structure (euchromatin) to a compact chromatin structure (heterochromatin) and vice versa
[8]. Histone acetyltransferase (HAT), histone deacetylase (HDAC), histone methyltransferase (HMT), and histone demethylase (HDM) enzymes interact specifically at methylated DNA regions, thus causing gene transcription or repression
[9][10].
1.3. Noncoding or MicroRNAs
Recently, it was discovered that noncoding RNAs are involved in gene regulation and genetic programming in both a healthy state and a CVD state
[11]. It was found that 98% of the human genome, which undergoes transcription without encoding for proteins, produces noncoding RNAs that are involved in important structural and regulatory functions. On the basis of size, these noncoding RNAs are classified into two types, i.e., long noncoding RNAs with a size of 0.2 kb to 2 kb and small noncoding RNAs, which consist of endogenous short interfering RNAs, PIWI-interacting RNAs, and microRNAs. Studies have also shown that these noncoding RNAs act as biomarkers of cardiovascular diseases (CVDs) and contribute to the pathogenesis of CVD
[12].
2. Cardiovascular Epigenetics
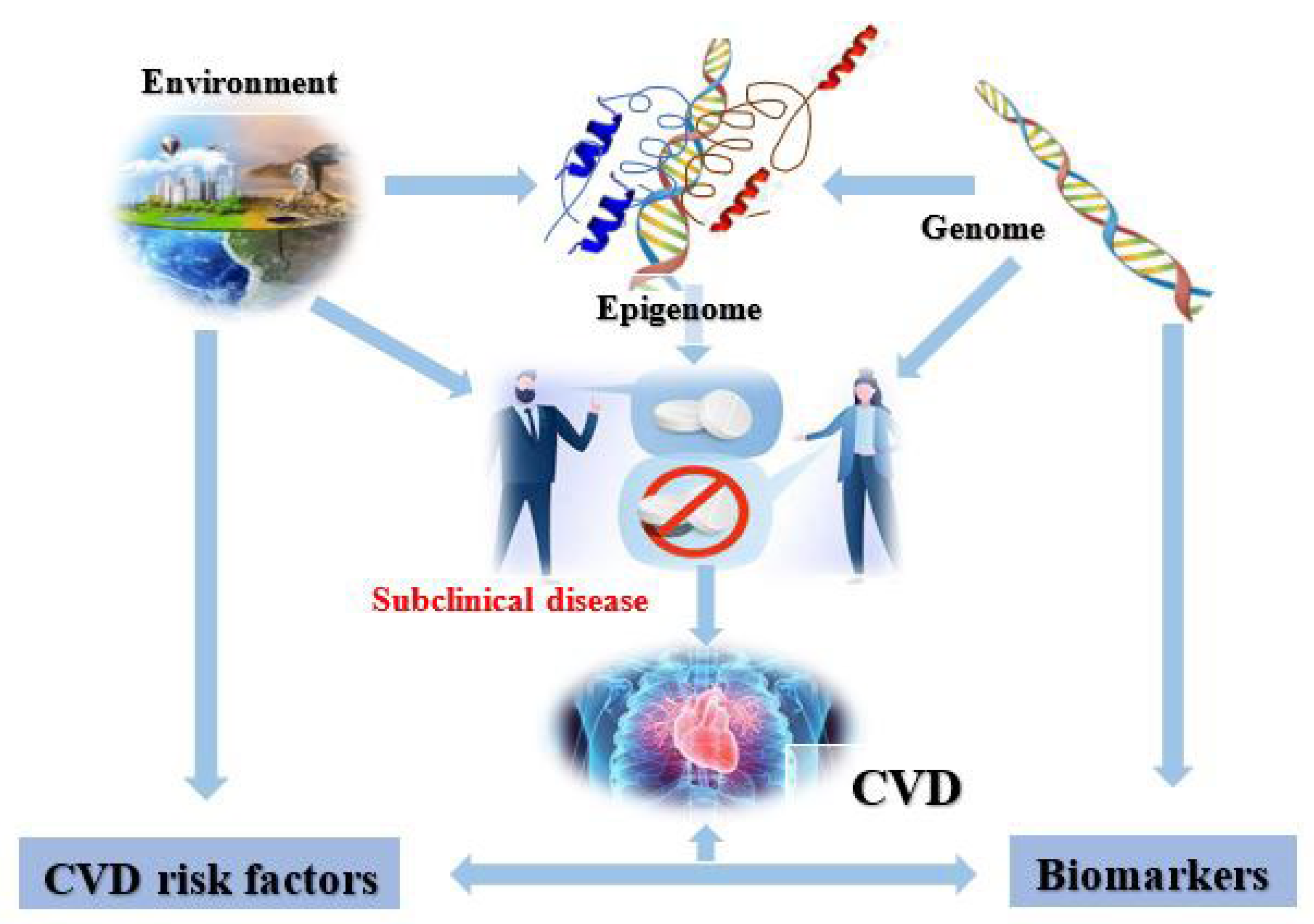
Epigenetics or epigenomics shows a critical association between phenotypic expression and genomic coding that is affected by both environmental and genetic factors (
Figure 2). Studies have shown that cardiovascular risk factors may affect and rearrange epigenetic patterns, and these cardiovascular biomarkers are said to be affiliated with epigenetic modifications. These epigenetic modifications are associated with clinical and subclinical cardiovascular diseases. Epigenetics is considered to be interconnected with genetics because these modifications (DNA methylation and histone modification) can change the expression of these genetic variations
[13].
Figure 2. Epigenomics to cardiovascular diseases and risk factors.
3. Techniques for Individual Epigenetic Mapping of Cardiovascular Disorders
In contrast with the belief that every individual has a somewhat similar genome, scientists proposed that all the epigenomes are not born same. Instead of an exception, diversity is considered to be a norm when it comes to the epigenetic modifications. When scientists completed the Human Genome Project, it became the cornerstone of genomic research
[14]. NGS, advances in bioinformatics, and GWAS helped in analyzing the increasing number of genomic datasets
[15]. However, despite these advances, the field of precision medicine in epigenomic cardiology remains uncharted. Although there are some barriers left to overcome, efforts have begun to complete the cardiovascular epigenome
[16].
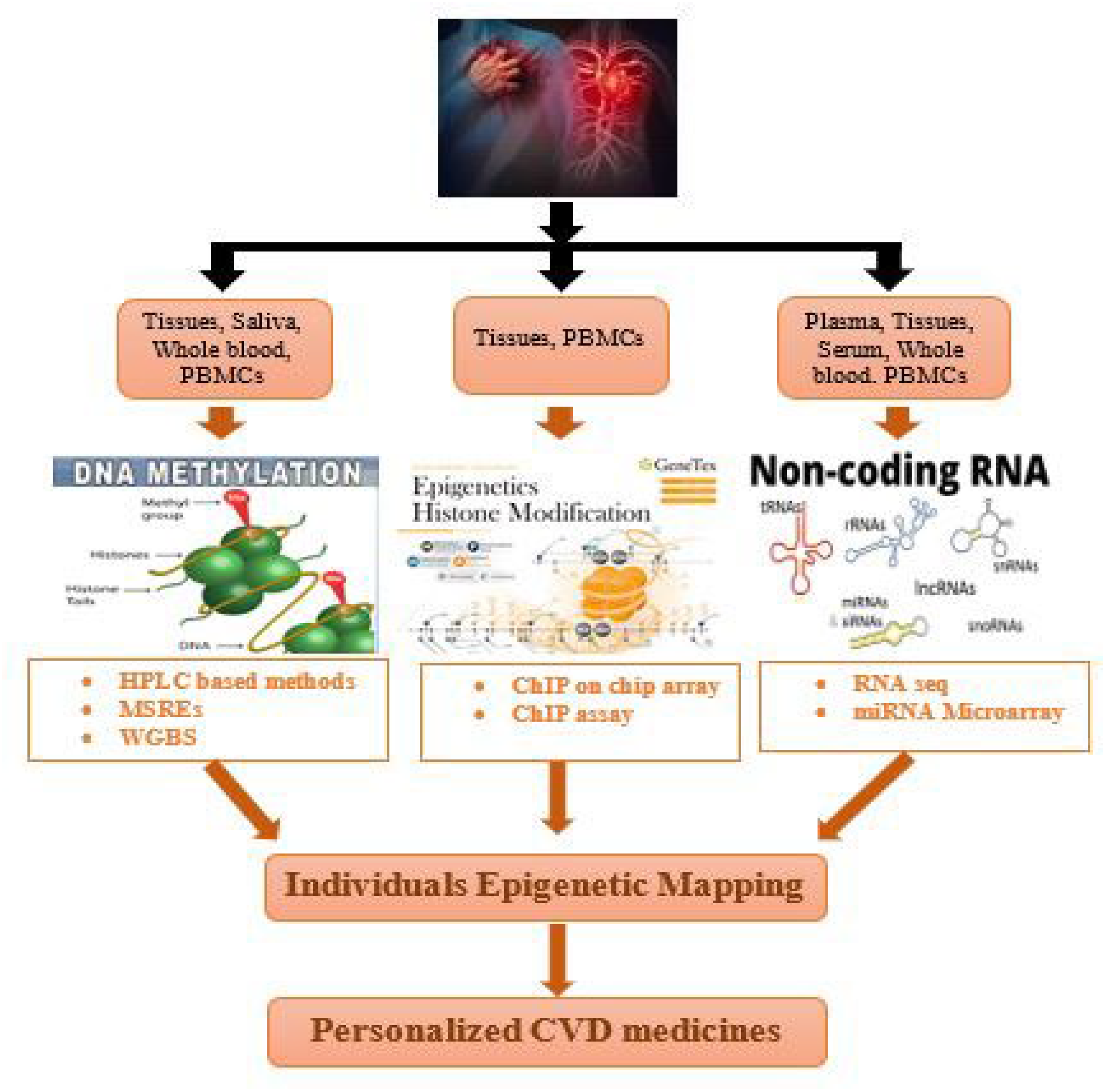
In order to enable clinical applications, various technological advancements have been introduced in the field of epigenomics. However, there still remains a key challenge for cheap performance of whole-genome bisulfite sequencing (WGBS) studies for EWAS (epigenome-wide association studies) (Table 1, Figure 3).
Figure 3. Application of epigenetic mapping techniques for CVD patients.
Table 1. Comparison of different scChIP seq methods
[17].
| Methods |
Strategy |
Mapping Rate |
Cell State |
Device for Cell Sorting |
| CUT and Tag |
ChIP-free, Tn5-barcoding (1 round) |
97% |
Native |
Costly Takara ICELL8 |
| Co-BATCH |
ChIP-free, Tn5-barcoding (for 2 rounds) |
94% |
Fixed and native |
FACS |
| sc-itChIP-seq |
ChIP and Tn5-barcoding (1 round) |
94% |
Fixed and native |
FACS |
| scDrop-ChIP |
Microfluidic system and ChIP for droplet formation |
70% |
Native |
Costly microfluidic device |
4. ChIP-on-Chip Guide
Chromatin immunoprecipitation (ChIP) followed by DNA microarray (chip), collectively called ChIP-on-chip, was the earliest technology used for large-scale epigenetic mapping, allowing the scientists to identify the protein–DNA interactions on a genome-wide level
[18]. It is based on the principle of DNA microchip hybridization, where a large number of probes covering a specific region or whole genome are seeded on a high-density chip. However, this technique suffered disadvantages such as signal bias, low resolution, inapplicability to a broad range of species, and ambiguous factors introduced by probe design
[19].
Chromatin immunoprecipitation sequencing (ChIP-seq) techniques, in comparison to ChIP-on-chip techniques, provides greater coverage, less noise, and higher resolution
[20][21]. Due to the fast decrease in the price of second-generation sequencing (SGS), ChIP seq has now become an essential technology in determining the epigenetics and gene regulation. This technology can also be used to determine enhancers, transcriptional factors, and various other regulatory elements
[22].
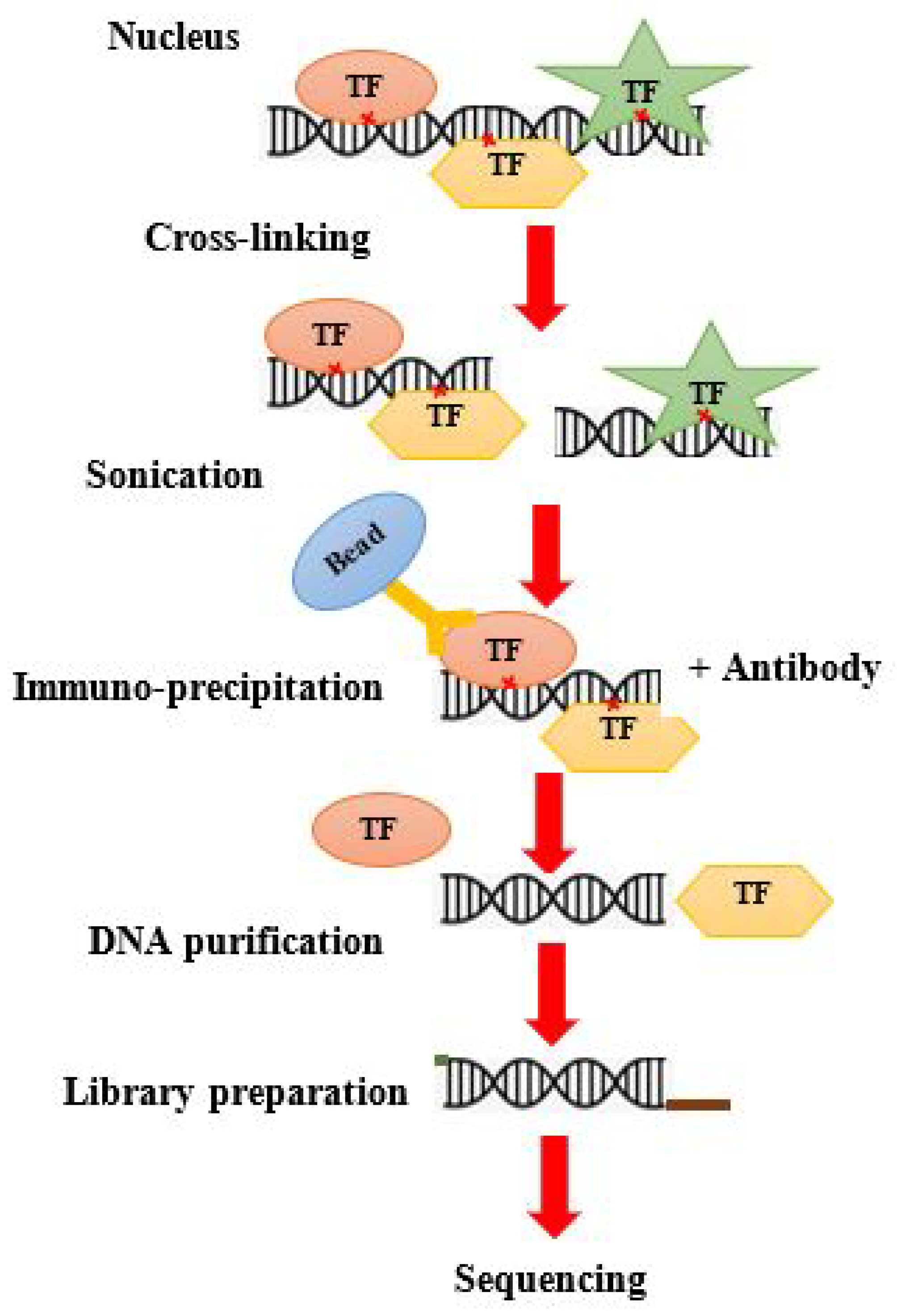
4.1. Traditional ChIP Seq
For protein–DNA complexed, ChIP seq procedures are performed to enrich the DNAs that are attached to specific proteins. It is a multistep experiment. At first, DNA is crosslinked with proteins via formaldehyde. Then, this crosslinked complex is subjected to sonication, which breaks it down into 200–600 bp small fragments. Then, the protein–DNA complex of interest is immunoprecipitated using an antibody against the respective protein. This releases the DNA, which is then subjected to DNA end repair, ligation of the adapter molecule, and construction of the library. Next, the required DNA is sequenced (
Figure 4)
[23].
Figure 4. Workflow of ChIP seq.
4.2. Single-Cell ChIP Seq
Traditional ChIP seq technology is unable to identify the chromatin signature of individual cells. For this purpose, a new technique was introduced that was able to determine genetic diversity in heterogenous cell populations and obtain knowledge regarding the evolution of tumor population; this technique is called single-cell ChIP (scChIP). Droplet-based scChIP (Drop ChIP) combines scDNA barcoding with a microfluidic device, allowing scientists to gain a comparatively low map coverage per cell
[24][25]. scChIP seq technology allows the clustering of cell populations on the basis of chromatin landscape diversity and determination of unique chromatin features of the population; for example, the decline in H3K27me3 biomarkers in some cells may lead to chemoresistance or can cause CVD
[17].
Table 1 shows the comparison of different ChIP seq methods and the mapping percentage.
5. Role of ChIP Seq in Determining Epigenetic Signature Underlying Cardiac Hypertrophy
One of the leading causes of mortality worldwide is heart failure (HF), which is frequently followed by a condition called cardiac hypertrophy, a condition in which there is an expression of genes that are only activated during the fetal stage and a repression of genes that need to be activated in adults
[26]. Although epigenetics is considered to be essential in regulating transcription, its role remains unknown in cardiac hypertrophy. Genome-wide association studies (GWASs) of histone 3 lysine-36 trimethylation (H3K36me3) and DNA methylation in normal hearts and cardiomyopathic hearts of humans divulged a wide range of epigenetic patterns
[27]. When idiopathic dilated cardiomyopathic patients were interrogated with cardiac methylome, differences in methylation were detected not only in heart disease-related pathways but also in genes of heart failure with yet unknown functions such as adenosine receptor A2A (ADORA2A) and lymphocyte antigen 75 (LY75)
[28]. High levels of miRNA-508-5p and miR-499 and low levels of circulating miRNA-342-3p, miRNA-30b, miRNA-142-3p, and miRNA-103 were detected in patients with advanced heart failure
[29][30]. Moreover, if low levels of miRNA-423-5p remain in these patients for a long time, it may result in bad outcomes
[31].
A study was conducted in adult mouse cardiomyocytes to describe epigenetic changes that occurred when they were subjected to a pro-hypertrophy stimulus in vivo. Genome-wide chromatin maps were generated and compared for the gene expression of normal and hypertrophic cardiomyocytes. These cardiomyocytes were isolated from the left ventricle of mouse hearts, which were subjected to transverse aortic constriction, and ChIP seq was performed using these cells with antibodies against active regulatory regions associated with three markers, i.e., H3K4me3, H3K27ac, and H3K9ac, as well as repressed regions represented by H3K27me3, H3K9me3, and H3K9me2
[32][33], and transcribed genes represented by H3K4me3
[34].
Overall, 9.1% of the genome of cardiac hypertrophic cells showed an alteration in the distribution of at least one histone mark was rearranged to transcriptional start sites (TSSs). Promoters of hypertrophic cardiomyocytes showed distinguished epigenetic patterns, and 9207 active enhancers were discovered with modulated activity. A role for myocyte enhancer factor (MEF)2C and MEF2A in regulating enhancers was identified by analyzing the transcriptional network within which the genetic elements tried to orchestrate hypertrophy gene expression
[35].
 +1 credit
+1 credit