 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Annarita Di Mise | -- | 4334 | 2022-06-27 19:36:32 | | | |
| 2 | Lindsay Dong | Meta information modification | 4334 | 2022-07-04 06:02:13 | | |
Video Upload Options
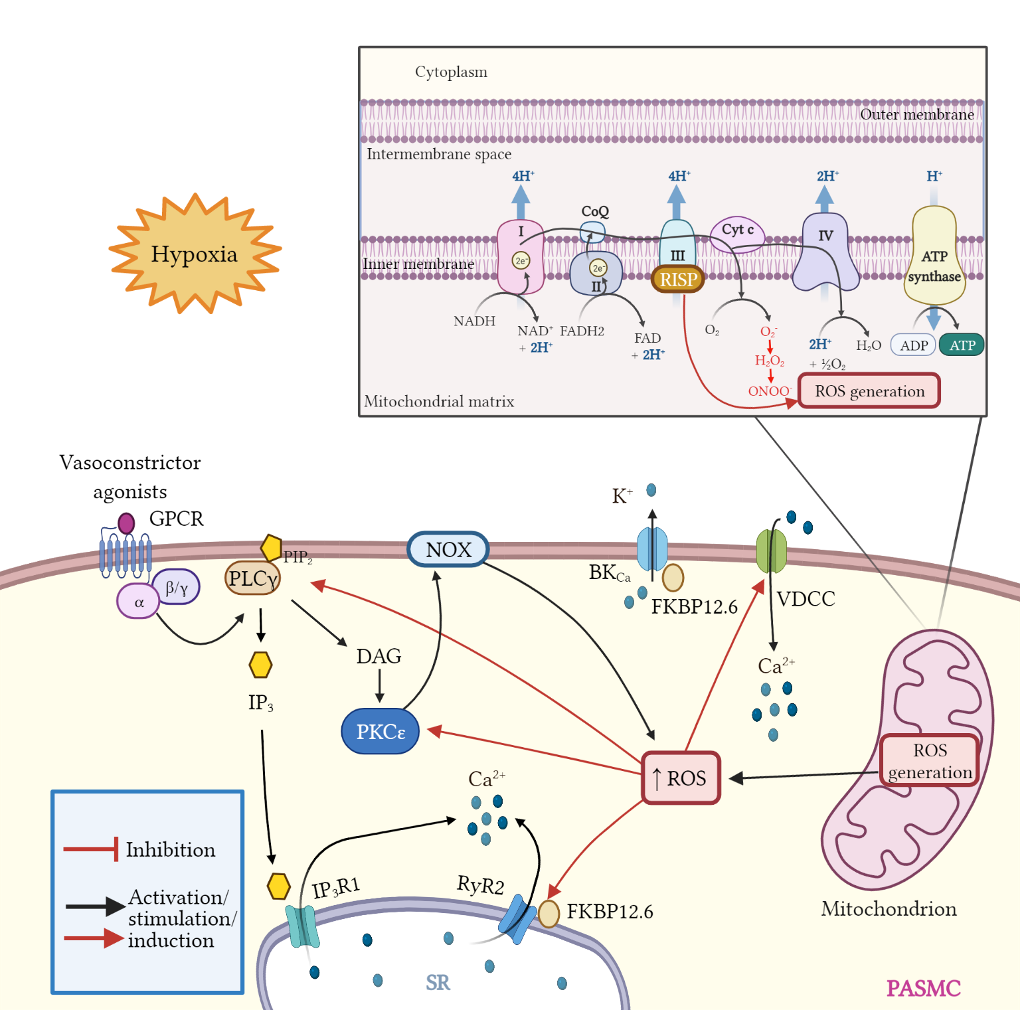
Mitochondria are important organelles that act as a primary site to produce reactive oxygen species (ROS). Additionally, mitochondria play a pivotal role in the regulation of Ca2+ signaling, fatty acid oxidation, and ketone synthesis. Dysfunction of these signaling molecules leads to the development of pulmonary hypertension (PH), atherosclerosis, and other vascular diseases. Features of PH include vasoconstriction and pulmonary artery (PA) remodeling, which can result from abnormal proliferation, apoptosis, and migration of PA smooth muscle cells (PASMCs). These responses are mediated by increased Rieske iron–sulfur protein (RISP)-dependent mitochondrial ROS production and increased mitochondrial Ca2+ levels. Mitochondrial ROS and Ca2+ can both synergistically activate nuclear factor κB (NF-κB) to trigger inflammatory responses leading to PH, right ventricular failure, and death.
1. Introduction
2. Pulmonary Hypertension
Table 1. WHO classification of pulmonary hypertension (PH).
|
WHO group |
Clinical classification |
Covering subtypes |
|
I |
Pulmonary arterial hypertension (PAH) |
Idiopathic; Drug and toxin-Induced; Heritable; Associated with connective tissue diseases, HIV infection, portal hypertension, schistosomiasis; PAH responder to Ca2+ channel blockers; Associated with pulmonary venous/capillaries occlusion; Persistent pulmonary hypertension of the newborn. |
|
II |
PH due to left heart diseases |
Heart failure; Valvular heart disease; Congenital or acquired cardiomyopathies; Failure with preserved/reduced ejection fraction. |
|
III |
PH due to lung disease or hypoxia |
COPD/hypoxia that includes COPD; Restrictive lung disease; Pulmonary disease with obstructive and restrictive pattern; Interstitial lung disease; Hypoxia without other lung diseases. |
|
VI |
PH due to the obstruction of pulmonary artery |
Chronic thromboembolic pulmonary hypertension (CTEPH); Other pulmonary artery obstructions. |
|
V |
PH due to unclear/multifactorial mechanisms |
Hematologic disorders; Metabolic disorders; Others. |
3. Inflammation in Pulmonary Hypertension
4. Mitochondria in Vascular Remodeling during PH
5. Mitochondrial ROS in Pulmonary Vasoconstriction and Endothelial Dysfunction
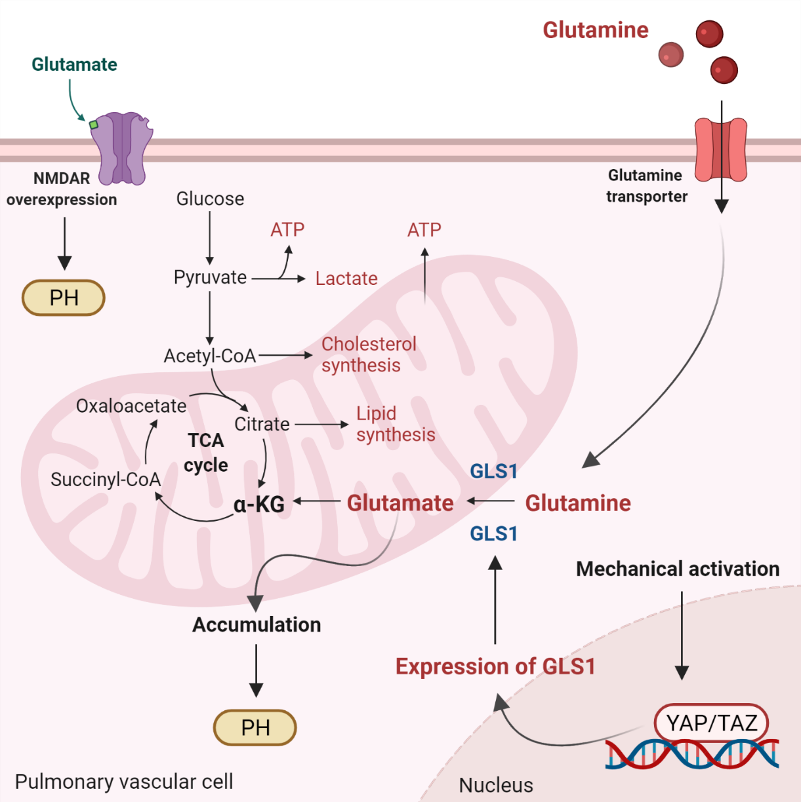
6. Mitochondrial Ca2+, ROS, and Glutaminolysis
Glutaminolysis is a mitochondrial process responsible for obtaining cellular energy from the deamination of glutamine to glutamate by glutaminase (GLS1) [139]. Subsequently, glutamate is converted to α-ketoglutarate (α-KG) by glutamate dehydrogenase. This process (anaplerotic reactions) helps to replenish the intermediates of the TCA cycle after they have been consumed and provides energy especially for proliferating cells. The increase in glutaminolysis leads to increased expression of GLS1 and increased uptake of glutamine by the pulmonary vasculature, resulting in increased glutamate production by pulmonary vascular cells and promoting PH. Activation of the former transcriptional coactivators triggers upregulation of GLS1 and leads to glutaminolysis, which maintains the hyperproliferative state and migration of pulmonary vascular cells in PH (Figure 2) [141].
7. Ketones and Mitochondrial Signaling
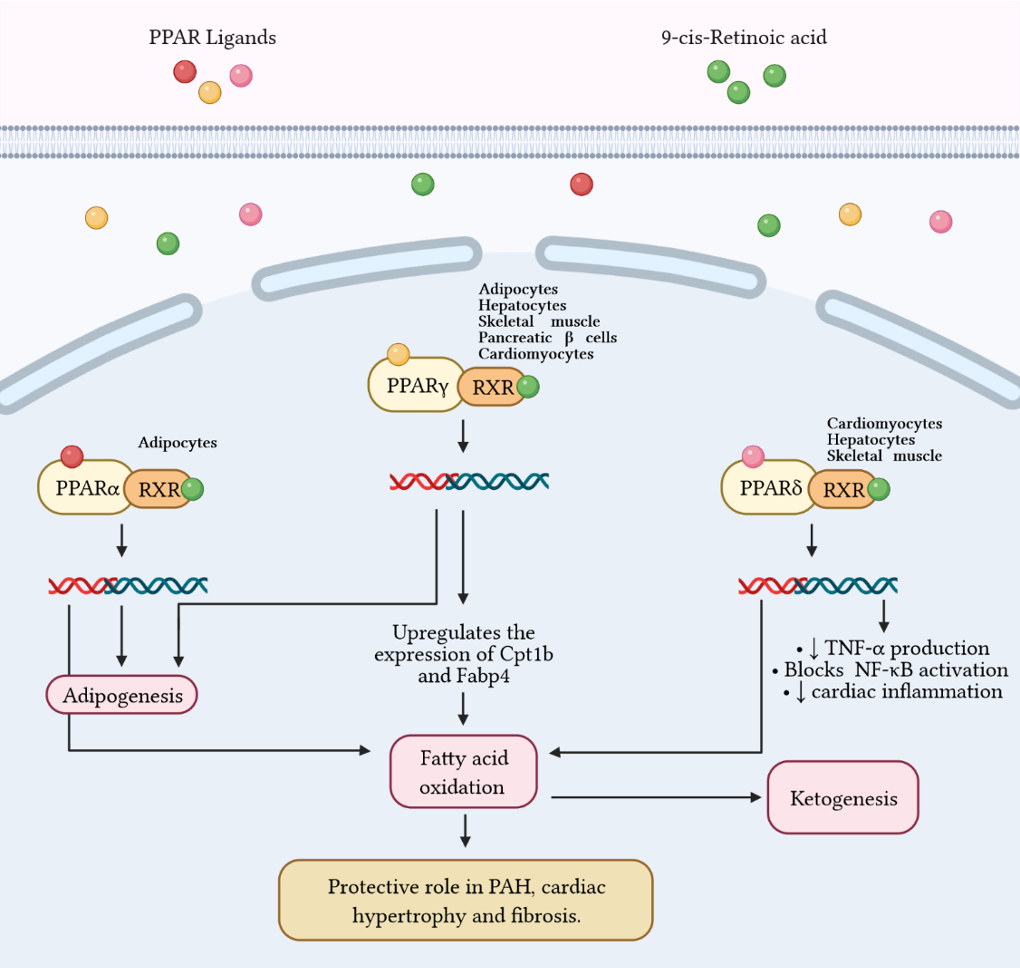
8. Conclusions
References
- Bruno, S.R.; Anathy, V. Lung epithelial endoplasmic reticulum and mitochondrial 3D ultrastructure: A new frontier in lung diseases. Histochem. Cell Biol. 2021, 155, 291–300.
- Bertram, R.; Pedersen, M.G.; Luciani, D.S.; Sherman, A. A simplified model for mitochondrial ATP production. J. Theor. Biol. 2006, 243, 575–586.
- Grumbach, I.M.; Nguyen, E.K. Metabolic Stress. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 991–997.
- Waypa, G.B.; Marks, J.D.; Guzy, R.D.; Mungai, P.T.; Schriewer, J.M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am. J. Respir. Crit. Care Med. 2013, 187, 424–432.
- Mei, L.; Zheng, Y.-M.; Song, T.; Yadav, V.R.; Joseph, L.C.; Truong, L.; Kandhi, S.; Barroso, M.M.; Takeshima, H.; Judson, M.A.; et al. Rieske iron-sulfur protein induces FKBP12.6/RyR2 complex remodeling and subsequent pulmonary hypertension through NF-κB/cyclin D1 pathway. Nat. Commun. 2020, 11, 3527.
- Rathore, R.; Zheng, Y.-M.; Niu, C.-F.; Liu, Q.-H.; Korde, A.; Ho, Y.-S.; Wang, Y.-X. Hypoxia activates NADPH oxidase to increase i and i through the mitochondrial ROS-PKCɛ signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 1223–1231.
- Truong, L.; Zheng, Y.-M.; Wang, Y.-X. Mitochondrial Rieske iron–sulfur protein in pulmonary artery smooth muscle: A key primary signaling molecule in pulmonary hypertension. Arch. Biochem. Biophys. 2020, 683, 108234.
- Yadav, V.R.; Song, T.; Mei, L.; Joseph, L.; Zheng, Y.-M.; Wang, Y.-X. PLCγ1-PKCε-IP3R1 signaling plays an important role in hypoxia-induced calcium response in pulmonary artery smooth muscle cells. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2018, 314, L724–L735.
- Yang, Z.; Song, T.; Truong, L.; Reyes-Garcia, J.; Wang, L.; Zheng, Y.-M.; Wang, Y.-X. Important role of sarcoplasmic reticulum Ca2+ release via ryanodine receptor-2 channel in hypoxia-induced rieske iron–sulfur protein-mediated mitochondrial reactive oxygen species generation in pulmonary artery smooth muscle cells. Antioxid. Redox Signal. 2020, 32, 447–462.
- Mohamed, R.; Dayati, P.; Mehr, R.N.; Kamato, D.; Seif, F.; Babaahmadi-Rezaei, H.; Little, P.J. Transforming growth factor–β1 mediated CHST11 and CHSY1 mRNA expression is ROS dependent in vascular smooth muscle cells. J. Cell Commun. Signal. 2019, 13, 225–233.
- Salazar, G.; Huang, J.; Feresin, R.; Zhao, Y.; Griendling, K. Zinc regulates Nox1 expression through a NF-κB and mitochondrial ROS dependent mechanism to induce senescence of vascular smooth muscle cells. Free Radic. Biol. Med. 2017, 108, 225–235.
- Pieczenik, S.R.; Neustadt, J. Mitochondrial dysfunction and molecular pathways of disease. Exp. Mol. Pathol. 2007, 83, 84–92.
- Chen, J.; Stimpson, S.; Fernandez-Bueno, G.A.; Mathews, C.E. Mitochondrial reactive oxygen species and type 1 diabetes. Antioxid. Redox Signal. 2018, 29, 1361–1372.
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285.
- Favero, G.; Bonomini, F.; Franco, C.; Rezzani, R. Mitochondrial dysfunction in skeletal muscle of a fibromyalgia model: The potential benefits of melatonin. Int. J. Mol. Sci. 2019, 20, 765.
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13.
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184.
- Cano Sanchez, M.; Lancel, S.; Boulanger, E.; Neviere, R. Targeting oxidative stress and mitochondrial dysfunction in the treatment of impaired wound healing: A systematic review. Antioxidants 2018, 7, 98.
- Zhu, Y.; Li, M.; Lu, Y.; Li, J.; Ke, Y.; Yang, J. Ilexgenin A inhibits mitochondrial fission and promote Drp1 degradation by Nrf2-induced PSMB5 in endothelial cells. Drug Dev. Res. 2019, 80, 481–489.
- Chen, J.; Wang, Y.; Dong, M.; Zhang, B.; Luo, Y.; Niu, W.; Li, Z. Reoxygenation reverses hypoxic pulmonary arterial remodeling by inducing smooth muscle cell apoptosis via reactive oxygen species–mediated mitochondrial dysfunction. J. Am. Heart Assoc. 2017, 6, e005602.
- Huetsch, J.C.; Suresh, K.; Shimoda, L.A. Regulation of smooth muscle cell proliferation by NADPH oxidases in pulmonary hypertension. Antioxidants 2019, 8, 56.
- Wang, M.; Luo, P.; Shi, W.; Guo, J.; Huo, S.; Yan, D.; Peng, L.; Zhang, C.; Lv, J.; Lin, L.; et al. S-nitroso-L-cysteine ameliorated pulmonary hypertension in the MCT-induced rats through anti-ROS and anti-inflammatory pathways. Oxidtive Med. Cell. Longev. 2021, 2021, 6621232.
- Zhuan, B.; Yu, Y.; Yang, Z.; Zhao, X.; Li, P. Mechanisms of oxidative stress effects of the NADPH oxidase-ROS-NF-kappaB transduction pathway and VPO1 on patients with chronic obstructive pulmonary disease combined with pulmonary hypertension. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3459–3464.
- Escribano-Subias, P.; Blanco, I.; Meseguer, M.L.; Lopez-Guarch, C.J.; Roman, A.; Morales, P.; Castillo-Palma, M.J.; Segovia, J.; Gómez-Sanchez, M.A.; Barberà, J.A. Survival in pulmonary hypertension in Spain: Insights from the Spanish registry. Eur. Respir. J. 2012, 40, 596–603.
- Ling, Y.; Johnson, M.K.; Kiely, D.G.; Condliffe, R.; Elliot, C.A.; Gibbs, J.S.R.; Howard, L.; Pepke-Zaba, J.; Sheares, K.K.K.; Corris, P.A.; et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: Results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am. J. Respir. Crit. Care Med. 2012, 186, 790–796.
- Peacock, A.J.; Murphy, N.F.; McMurray, J.J.V.; Caballero, L.; Stewart, S. An epidemiological study of pulmonary arterial hypertension. Eur. Respir. J. 2007, 30, 104–109.
- Wijeratne, D.T.; Lajkosz, K.; Brogly, S.B.; Lougheed, M.D.; Jiang, L.; Housin, A.; Barber, D.; Johnson, A.; Doliszny, K.M.; Archer, S.L. Increasing incidence and prevalence of World Health Organization groups 1 to 4 pulmonary hypertension: A population-based cohort study in Ontario, Canada. Circ. Cardiovasc. Qual. Outcomes 2018, 11, e003973.
- Prasad, K. AGE–RAGE Stress in the pathophysiology of pulmonary hypertension and its treatment. Int. J. Angiol. 2019, 28, 71–79.
- Hoeper, M.M.; Huscher, D.; Ghofrani, H.A.; Delcroix, M.; Distler, O.; Schweiger, C.; Grunig, E.; Staehler, G.; Rosenkranz, S.; Halank, M.; et al. Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: Results from the COMPERA registry. Int. J. Cardiol. 2013, 168, 871–880.
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.-F.; Chabot, F.; et al. Pulmonary arterial hypertension in France: Results from a national registry. Am. J. Respir. Crit. Care Med. 2006, 173, 1023–1030.
- Benza, R.L.; Gomberg-Maitland, M.; Miller, D.P.; Frost, A.; Frantz, R.P.; Foreman, A.J.; Badesch, D.B.; McGoon, M.D. The REVEAL registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest 2012, 141, 354–362.
- Jacobs, W.; van de Veerdonk, M.C.; Trip, P.; Man, F.H.-D.; Heymans, M.W.; Marcus, J.T.; Kawut, S.M.; Bogaard, H.-J.; Boonstra, A.; Noordegraaf, A.V. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest 2014, 145, 1230–1236.
- Sakuma, M.; Toyoda, S.; Inoue, T.; Node, K. Inflammation in pulmonary artery hypertension. Vasc. Pharmacol. 2019, 118–119, 106562.
- Singh, I.; Oliveira, R.; Naeije, R.; Rahaghi, F.N.; Oldham, W.M.; Systrom, D.M.; Waxman, A.B. Pulmonary vascular distensibility and early pulmonary vascular remodeling in pulmonary hypertension. Chest 2019, 156, 724–732.
- Giaid, A.; Saleh, D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1995, 333, 214–221.
- Kaneko, F.T.; Arroliga, A.C.; Dweik, R.A.; Comhair, S.A.; Laskowski, D.; Oppedisano, R.; Thomassen, M.J.; Erzurum, S.C. Biochemical reaction products of nitric oxide as quantitative markers of primary pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1998, 158, 917–923.
- Tuder, R.M.; Cool, C.D.; Geraci, M.W.; Wang, J.; Abman, S.H.; Wright, L.; Badesch, D.; Voelkel, N.F. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1999, 159, 1925–1932.
- Falcetti, E.; Hall, S.M.; Phillips, P.G.; Patel, J.; Morrell, N.W.; Haworth, S.G.; Clapp, L.H. Smooth muscle proliferation and role of the prostacyclin (IP) receptor in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2010, 182, 1161–1170.
- Humbert, M.; Montani, D.; Perros, F.; Dorfmüller, P.; Adnot, S.; Eddahibi, S. Endothelial cell dysfunction and cross talk between endothelium and smooth muscle cells in pulmonary arterial hypertension. Vasc. Pharmacol. 2008, 49, 113–118.
- Wilson, J.L.; Warburton, R.; Taylor, L.; Toksoz, D.; Hill, N.; Polgar, P. Unraveling endothelin-1 induced hypercontractility of human pulmonary artery smooth muscle cells from patients with pulmonary arterial hypertension. PLoS ONE 2018, 13, e0195780.
- Wang, X.-Y.; Mo, D.; Tian, W.; Liu, X.-X.; Zhou, Y.-G.; Sun, Y.; Feng, Y.-D.; Xiao, X.; Hao, X.-W.; Zhang, H.-N.; et al. Inhibition of RhoA/ROCK signaling pathway ameliorates hypoxic pulmonary hypertension via HIF-1α-dependent functional TRPC channels. Toxicol. Appl. Pharmacol. 2019, 369, 60–72.
- Guo, S.; Shen, Y.; He, G.; Wang, T.; Xu, D.; Wen, F. Involvement of Ca2+-activated K+ channel 3.1 in hypoxia-induced pulmonary arterial hypertension and therapeutic effects of TRAM-34 in rats. Biosci. Rep. 2017, 37, BSR20170763.
- Lee, H.; Kim, K.C.; Hong, Y.M. Change of voltage-gated potassium channel 1.7 expressions in monocrotaline-induced pulmonary arterial hypertension rat model. Korean J. Pediatr. 2018, 61, 271–278.
- Tuder, R.M. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res. 2017, 367, 643–649.
- George, M.P.; Champion, H.C.; Simon, M.; Guyach, S.; Tarantelli, R.; Kling, H.M.; Brower, A.; Janssen, C.; Murphy, J.; Carney, J.P.; et al. Physiologic changes in a nonhuman primate model of HIV-associated pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 48, 374–381.
- Graham, B.B.; Bandeira, A.P.; Morrell, N.W.; Butrous, G.; Tuder, R.M. Schistosomiasis-associated pulmonary hypertension: Pulmonary vascular disease: The global perspective. Chest 2010, 137, 20S–29S.
- McMahan, Z.H.; Hummers, L.K. Systemic sclerosis—Challenges for clinical practice. Nat. Rev. Rheumatol. 2013, 9, 90–100.
- Li, K.; Li, Y.; Yu, Y.; Ding, J.; Huang, H.; Chu, C.; Hu, L.; Yu, Y.; Cao, Y.; Xu, P.; et al. Bmi-1 alleviates adventitial fibroblast senescence by eliminating ROS in pulmonary hypertension. BMC Pulm. Med. 2021, 21, 80.
- Li, X.; Hu, B.; Wang, L.; Xia, Q.; Ni, X. P2X7 receptor-mediated phenotype switching of pulmonary artery smooth muscle cells in hypoxia. Mol. Biol. Rep. 2021, 48, 2133–2142.
- Zhu, L.; Liu, F.; Hao, Q.; Feng, T.; Chen, Z.; Luo, S.; Xiao, R.; Sun, M.; Zhang, T.; Fan, X.; et al. Dietary geranylgeranyl pyrophosphate counteracts the benefits of statin therapy in experimental pulmonary hypertension. Circulation 2021, 143, 1775–1792.
- Bueno-Beti, C.; Hadri, L.; Hajjar, R.J.; Sassi, Y. The Sugen 5416/Hypoxia mouse model of pulmonary arterial hypertension. In Experimental Models of Cardiovascular Diseases; Humana Press: New York, NY, USA, 2018; Volume 1816, pp. 243–252.
- Morciano, G.; Vitto, V.; Bouhamida, E.; Giorgi, C.; Pinton, P. Mitochondrial bioenergetics and dynamism in the failing heart. Life 2021, 11, 436.
- Mey, J.T.; Hari, A.; Axelrod, C.L.; Fealy, C.E.; Erickson, M.L.; Kirwan, J.P.; Dweik, R.A.; Heresi, G.A. Lipids and ketones dominate metabolism at the expense of glucose control in pulmonary arterial hypertension: A hyperglycaemic clamp and metabolomics study. Eur. Respir. J. 2020, 55, 1901700.
- Groth, A.; Vrugt, B.; Brock, M.; Speich, R.; Ulrich, S.; Huber, L.C. Inflammatory cytokines in pulmonary hypertension. Respir. Res. 2014, 15, 47.
- Fujita, M.; Mason, R.J.; Cool, C.; Shannon, J.M.; Hara, N.; Fagan, K.A. Pulmonary hypertension in TNF-α-overexpressing mice is associated with decreased VEGF gene expression. J. Appl. Physiol. 2002, 93, 2162–2170.
- Humbert, M.; Monti, G.; Brenot, F.; Sitbon, O.; Portier, A.; Grangeot-Keros, L.; Duroux, P.; Galanaud, P.; Simonneau, G.; Emilie, D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1995, 151, 1628–1631.
- Barzilai, S.; Blecher-Gonen, R.; Barnett-Itzhaki, Z.; Zauberman, A.; Lebel-Haziv, Y.; Amit, I.; Alon, R. M-sec regulates polarized secretion of inflammatory endothelial chemokines and facilitates CCL2-mediated lymphocyte transendothelial migration. J. Leukoc. Biol. 2016, 99, 1045–1055.
- Voelkel, N.F.; Tuder, R.M.; Bridges, J.; Arend, W.P. Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am. J. Respir. Cell Mol. Biol. 1994, 11, 664–675.
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927.
- Pak, O.; Sommer, N.; Hoeres, T.; Bakr, A.; Waisbrod, S.; Sydykov, A.; Haag, D.; Esfandiary, A.; Kojonazarov, B.; Veit, F.; et al. Mitochondrial hyperpolarization in pulmonary vascular remodeling. Mitochondrial uncoupling protein deficiency as disease model. Am. J. Respir. Cell Mol. Biol. 2013, 49, 358–367.
- Sommer, N.; Strielkov, I.; Pak, O.; Weissmann, N. Oxygen sensing and signal transduction in hypoxic pulmonary vasoconstriction. Eur. Respir. J. 2016, 47, 288–303.
- Weir, E.K.; Archer, S.L. The role of redox changes in oxygen sensing. Respir. Physiol. Neurobiol. 2010, 174, 182–191.
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105.
- Yang, M.; Dart, C.; Kamishima, T.; Quayle, J.M. Hypoxia and metabolic inhibitors alter the intracellular ATP:ADP ratio and membrane potential in human coronary artery smooth muscle cells. PeerJ 2020, 8, e10344.
- Xu, W.; Janocha, A.J.; Erzurum, S.C. Metabolism in pulmonary hypertension. Annu. Rev. Physiol. 2021, 83, 551–576.
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270.
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314.
- Boehme, J.; Sun, X.; Tormos, K.V.; Gong, W.; Kellner, M.; Datar, S.A.; Kameny, R.J.; Yuan, J.X.-J.; Raff, G.W.; Fineman, J.R.; et al. Pulmonary artery smooth muscle cell hyperproliferation and metabolic shift triggered by pulmonary overcirculation. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H944–H957.
- Dabral, S.; Tian, X.; Kojonazarov, B.; Savai, R.; Ghofrani, H.A.; Weissmann, N.; Florio, M.; Sun, J.; Jonigk, D.; Maegel, L.; et al. Notch1 signalling regulates endothelial proliferation and apoptosis in pulmonary arterial hypertension. Eur. Respir. J. 2016, 48, 1137–1149.
- Xu, S.; Xu, X.; Zhang, J.; Ying, K.; Shao, Y.; Zhang, R. Pulmonary hypertension as a manifestation of mitochondrial disease: A case report and review of the literature. Medicine 2017, 96, e8716.
- Thistlethwaite, P.A. Linking vascular remodeling and inflammation in pulmonary arterial hypertension: Is there a common root cause? Am. J. Respir. Cell Mol. Biol. 2017, 57, 15–17.
- Kuznetsov, A.V.; Margreiter, R.; Amberger, A.; Saks, V.; Grimm, M. Changes in mitochondrial redox state, membrane potential and calcium precede mitochondrial dysfunction in doxorubicin-induced cell death. Biochim. Biophys. Acta 2011, 1813, 1144–1152.
- Bonnet, S.; Michelakis, E.D.; Porter, C.; Andrade, M.; Thébaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; et al. An abnormal mitochondrial–hypoxia inducible factor-1α–Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats. Circulation 2006, 113, 2630–2641.
- Bonnet, S.; Rochefort, G.; Sutendra, G.; Archer, S.L.; Haromy, A.; Webster, L.; Hashimoto, K.; Michelakis, E.D. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc. Natl. Acad. Sci. USA 2007, 104, 11418–11423.
- McMurtry, M.S.; Archer, S.L.; Altieri, D.C.; Bonnet, S.; Haromy, A.; Harry, G.; Bonnet, S.; Puttagunta, L.; Michelakis, E.D. Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J. Clin. Investig. 2005, 115, 1479–1491.
- Hu, H.-L.; Zhang, Z.-X.; Chen, C.-S.; Cai, C.; Zhao, J.-P.; Wang, X. Effects of mitochondrial potassium channel and membrane potential on hypoxic human pulmonary artery smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 2010, 42, 661–666.
- Gaudry, M.J.; Jastroch, M. Molecular evolution of uncoupling proteins and implications for brain function. Neurosci. Lett. 2019, 696, 140–145.
- Boucherat, O.; Peterlini, T.; Bourgeois, A.; Nadeau, V.; Breuils-Bonnet, S.; Boilet-Molez, S.; Potus, F.; Meloche, J.; Chabot, S.; Lambert, C.; et al. Mitochondrial HSP90 accumulation promotes vascular remodeling in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 90–103.
- Sobolewski, A.; Rudarakanchana, N.; Upton, P.D.; Yang, J.; Crilley, T.K.; Trembath, R.; Morrell, N. Failure of bone morphogenetic protein receptor trafficking in pulmonary arterial hypertension: Potential for rescue. Hum. Mol. Genet. 2008, 17, 3180–3190.
- Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Sutendra, G.; Michelakis, E.D. Attenuating endoplasmic reticulum stress as a novel therapeutic strategy in pulmonary hypertension. Circulation 2013, 127, 115–125.
- Guignabert, C.; Bailly, S.; Humbert, M. Restoring BMPRII functions in pulmonary arterial hypertension: Opportunities, challenges and limitations. Expert Opin. Ther. Targets 2017, 21, 181–190.
- Zhuan, B.; Wang, X.; Wang, M.-D.; Li, Z.-C.; Yuan, Q.; Xie, J.; Yang, Z. Hypoxia induces pulmonary artery smooth muscle dysfunction through mitochondrial fragmentation-mediated endoplasmic reticulum stress. Aging 2020, 12, 23684–23697.
- Lindner, P.; Christensen, S.B.; Nissen, P.; Møller, J.V.; Engedal, N. Cell death induced by the ER stressor thapsigargin involves death receptor 5, a non-autophagic function of MAP1LC3B, and distinct contributions from unfolded protein response components. Cell Commun. Signal. 2020, 18, 12.
- Shore, G.C.; Papa, F.R.; Oakes, S.A. Signaling cell death from the endoplasmic reticulum stress response. Curr. Opin. Cell Biol. 2011, 23, 143–149.
- Yang, Y.-D.; Li, M.-M.; Xu, G.; Zhang, E.-L.; Chen, J.; Sun, B.; Chen, D.-W.; Gao, Y.-Q. Targeting mitochondria-associated membranes as a potential therapy against endothelial injury induced by hypoxia. J. Cell. Biochem. 2019, 120, 18967–18978.
- Siques, P.; Brito, J.; Pena, E. Reactive oxygen species and pulmonary vasculature during hypobaric hypoxia. Front. Physiol. 2018, 9, 865.
- Waypa, G.B.; Guzy, R.; Mungai, P.T.; Mack, M.M.; Marks, J.D.; Roe, M.W.; Schumacker, P.T. Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ. Res. 2006, 99, 970–978.
- Dunham-Snary, K.; Wu, D.; Potus, F.; Sykes, E.A.; Mewburn, J.D.; Charles, R.L.; Eaton, P.; Sultanian, R.A.; Archer, S.L. Ndufs2, a core subunit of mitochondrial complex I, is essential for acute oxygen-sensing and hypoxic pulmonary vasoconstriction. Circ. Res. 2019, 124, 1727–1746.
- Dunham-Snary, K.; Wu, D.; Sykes, E.A.; Thakrar, A.; Parlow, L.R.; Mewburn, J.D.; Parlow, J.L.; Archer, S.L. Hypoxic pulmonary vasoconstriction: From molecular mechanisms to medicine. Chest 2017, 151, 181–192.
- Li, X.-Q.; Zheng, Y.-M.; Reyes-García, J.; Wang, Y.-X. Diversity of ryanodine receptor 1-mediated Ca2+ signaling in systemic and pulmonary artery smooth muscle cells. Life Sci. 2021, 270, 119016.
- Liao, B.; Zheng, Y.-M.; Yadav, V.R.; Korde, A.S.; Wang, Y.-X. Hypoxia induces intracellular Ca2+ release by causing reactive oxygen species-mediated dissociation of FK506-binding protein 12.6 from ryanodine receptor 2 in pulmonary artery myocytes. Antioxid. Redox Signal. 2011, 14, 37–47.
- Wang, Y.-X.; Zheng, Y.-M. Role of ROS signaling in differential hypoxic Ca2+ and contractile responses in pulmonary and systemic vascular smooth muscle cells. Respir. Physiol. Neurobiol. 2010, 174, 192–200.
- Wang, Y.-X.; Zheng, Y.-M.; Mei, Q.-B.; Wang, Q.-S.; Collier, M.L.; Fleischer, S.; Xin, H.-B.; Kotlikoff, M.I. FKBP12.6 and cADPR regulation of Ca2+ release in smooth muscle cells. Am. J. Physiol. Cell Physiol. 2004, 286, C538–C546.
- Tang, W.-X.; Chen, Y.-F.; Zou, A.-P.; Campbell, W.B.; Li, P.-L. Role of FKBP12.6 in cADPR-induced activation of reconstituted ryanodine receptors from arterial smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1304–H1310.
- Korde, A.; Yadav, V.R.; Zheng, Y.-M.; Wang, Y.-X. Primary role of mitochondrial Rieske iron–sulfur protein in hypoxic ROS production in pulmonary artery myocytes. Free Radic. Biol. Med. 2011, 50, 945–952.
- Song, T.; Zheng, Y.-M.; Wang, Y.-X. Cross talk between mitochondrial reactive oxygen species and sarcoplasmic reticulum calcium in pulmonary arterial smooth muscle cells. Adv. Exp. Med. Biol. 2017, 967, 289–298.
- Wang, Y.-X.; Zheng, Y.-M. ROS-Dependent signaling mechanisms for hypoxic Ca2+ responses in pulmonary artery myocytes. Antioxid. Redox Signal. 2010, 12, 611–623.
- Grabacka, M.; Pierzchalska, M.; Dean, M.; Reiss, K. Regulation of ketone body metabolism and the role of PPARα. Int. J. Mol. Sci. 2016, 17, 2093.
- Nasser, S.; Vialichka, V.; Biesiekierska, M.; Balcerczyk, A.; Pirola, L. Effects of ketogenic diet and ketone bodies on the cardiovascular system: Concentration matters. World J. Diabetes 2020, 11, 584–595.
- Puchalska, P.; Crawford, P.A. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 2017, 25, 262–284.
- De Sa, H.A.; Chung, S.; Shaniuk, P.M. Sweet and salty: Diabetic ketoacidosis in a patient with nephrogenic diabetes insipidus. Cureus 2021, 13, e12682.
- Gibson, A.A.; Eroglu, E.I.; Rooney, K.; Harper, C.; McClintock, S.; Franklin, J.; Markovic, T.P.; Seimon, R.V.; Sainsbury, A. Urine dipsticks are not accurate for detecting mild ketosis during a severely energy restricted diet. Obes. Sci. Pract. 2020, 6, 544–551.
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52.
- Arima, Y.; Nakagawa, Y.; Takeo, T.; Ishida, T.; Yamada, T.; Hino, S.; Nakao, M.; Hanada, S.; Umemoto, T.; Suda, T.; et al. Murine neonatal ketogenesis preserves mitochondrial energetics by preventing protein hyperacetylation. Nat. Metab. 2021, 3, 196–210.
- Dhillon, K.K.; Gupta, S. Biochemistry, ketogenesis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–426.
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose tissue remodeling: Its role in energy metabolism and metabolic disorders. Front. Endocrinol. 2016, 7, 30.
- Kadochi, Y.; Mori, S.; Fujiwara-Tani, R.; Luo, Y.; Nishiguchi, Y.; Kishi, S.; Fujii, K.; Ohmori, H.; Kuniyasu, H. Remodeling of energy metabolism by a ketone body and medium-chain fatty acid suppressed the proliferation of CT26 mouse colon cancer cells. Oncol. Lett. 2017, 14, 673–680.
- Serviddio, G.; Giudetti, A.M.; Bellanti, F.; Priore, P.; Rollo, T.; Tamborra, R.; Siculella, L.; Vendemiale, G.; Altomare, E.; Gnoni, G.V. Oxidation of hepatic carnitine palmitoyl transferase-I (CPT-I) impairs fatty acid beta-oxidation in rats fed a methionine-choline deficient diet. PLoS ONE 2011, 6, e24084.
- Latruffe, N. Human peroxisomal 3-ketoacyl-CoA thiolase: Tissue expression and metabolic regulation: Human peroxisomal thiolase. Adv. Exp. Med. Biol. 2020, 1299, 161–167.
- Galluzzi, L.; Kroemer, G. Aberrant ketolysis fuels hepatocellular cancer progression. Cell Res. 2016, 26, 1077–1078.
- Dumbrepatil, A.B.; Zegalia, K.A.; Sajja, K.; Kennedy, R.T.; Marsh, E.N.G. Targeting viperin to the mitochondrion inhibits the thiolase activity of the trifunctional enzyme complex. J. Biol. Chem. 2020, 295, 2839–2849.
- Fletcher, J.A.; Deja, S.; Satapati, S.; Fu, X.; Burgess, S.C.; Browning, J.D. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 2019, 5, e127737.
- Likhodii, S.S.; Musa, K.; Cunnane, S.C. Breath acetone as a measure of systemic ketosis assessed in a rat model of the ketogenic diet. Clin. Chem. 2002, 48, 115–120.
- Musa-Veloso, K.; Likhodii, S.S.; Cunnane, S.C. Breath acetone is a reliable indicator of ketosis in adults consuming ketogenic meals. Am. J. Clin. Nutr. 2002, 76, 65–70.
- Chen, F.; Qian, L.-H.; Deng, B.; Liu, Z.-M.; Zhao, Y.; Le, Y.-Y. Resveratrol protects vascular endothelial cells from high glucose-induced apoptosis through inhibition of NADPH oxidase activation-driven oxidative stress. CNS Neurosci. Ther. 2013, 19, 675–681.
- Zhu, M.; Chen, J.; Jiang, H.; Miao, C. Propofol protects against high glucose-induced endothelial adhesion molecules expression in human umbilical vein endothelial cells. Cardiovasc. Diabetol. 2013, 12, 13.
- Grinnan, D.; Farr, G.; Fox, A.; Sweeney, L. The role of hyperglycemia and insulin resistance in the development and progression of pulmonary arterial hypertension. J. Diabetes Res. 2016, 2016, 2481659.
- La Frano, M.R.; Fahrmann, J.F.; Grapov, D.; Pedersen, T.L.; Newman, J.; Fiehn, O.; Underwood, M.A.; Mestan, K.K.; Steinhorn, R.H.; Wedgwood, S. Umbilical cord blood metabolomics reveal distinct signatures of dyslipidemia prior to bronchopulmonary dysplasia and pulmonary hypertension. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2018, 315, L870–L881.
- Kim, D.; Roberts, C.; McKenzie, A.; George, M.P. Nutritional ketosis to treat pulmonary hypertension associated with obesity and metabolic syndrome: A case report. Pulm. Circ. 2021, 11, 2045894021991426.
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724.
- Kim, H.-I.; Ahn, Y.-H. Role of peroxisome proliferator-activated receptor-γ in the glucose-sensing apparatus of liver and β-cells. Diabetes 2004, 53, S60–S65.
- Semple, R.K.; Chatterjee, V.K.K.; O’Rahilly, S. PPAR gamma and human metabolic disease. J. Clin. Investig. 2006, 116, 581–589.
- Wang, Y.-X. PPARs: Diverse regulators in energy metabolism and metabolic diseases. Cell Res. 2010, 20, 124–137.
- Yang, Q.; Long, Q. PPARδ, a potential therapeutic target for heart disease. Nucl. Recept. Res. 2018, 5, 101375.
- Hansmann, G.; Calvier, L.; Risbano, M.G.; Chan, S.Y. Activation of the metabolic master regulator PPARγ: A potential pioneering therapy for pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2020, 62, 143–156.
- Green, D.E.; Murphy, T.C.; Kang, B.-Y.; Searles, C.D.; Hart, C.M. PPARγ ligands attenuate hypoxia-induced proliferation in human pulmonary artery smooth muscle cells through modulation of microRNA-21. PLoS ONE 2015, 10, e0133391.
- Reddy, A.; Lakshmi, S.; Kleinhenz, J.M.; Sutliff, R.L.; Hart, C.M.; Reddy, R.C. Endothelial cell peroxisome proliferator–activated receptor γ reduces endotoxemic pulmonary inflammation and injury. J. Immunol. 2012, 189, 5411–5420.

