Mitochondria are important organelles that act as a primary site to produce reactive oxygen species (ROS). Additionally, mitochondria play a pivotal role in the regulation of Ca2+ signaling, fatty acid oxidation, and ketone synthesis. Dysfunction of these signaling molecules leads to the development of pulmonary hypertension (PH), atherosclerosis, and other vascular diseases. Features of PH include vasoconstriction and pulmonary artery (PA) remodeling, which can result from abnormal proliferation, apoptosis, and migration of PA smooth muscle cells (PASMCs). These responses are mediated by increased Rieske iron–sulfur protein (RISP)-dependent mitochondrial ROS production and increased mitochondrial Ca2+ levels. Mitochondrial ROS and Ca2+ can both synergistically activate nuclear factor κB (NF-κB) to trigger inflammatory responses leading to PH, right ventricular failure, and death.
- pulmonary hypertension
- mitochondrial ROS
- ketones
- calcium signalling
1. Introduction
2. Pulmonary Hypertension
PH is a rare and fatal disease with an estimated prevalence of 5 to 50 cases per million individuals [29,30,31,32][24][25][26][27]. This ailment has been defined by the increase in the mean pulmonary arterial pressure (mPAP) ≥25 mm Hg at rest or ≥30 mm Hg during/after workout [33][28]. Most patients suffering from PH are women, covering around 60% to 80% of all cases [30,34,35][25][29][30]. Therefore, female sex has been considered a risk factor for the development of PH. Nevertheless, male sex has been associated with poorer survival rates and this could be explained by the fact that men affected by this illness exhibit a low right ventricular function recovery [36,37][31][32]. The latest update in 2013 of World Health Organization’s clinical classification system has catalogued PH into five categories/groups depending on the main underlying cause, hemodynamics, clinical features, and therapeutic responsiveness [21,33][21][28]. As shown in Table 1, Group I refers to pulmonary arterial hypertension (PAH) and encloses idiopathic PH (IPAH), drug-induced PH, heritable PH, and PH associated with other systemic diseases. PH due to left heart disease corresponds to group II. Groups III and IV include PH due to lung diseases and PA obstructions, respectively. Lastly, group V describes PH with unclear multifactorial mechanisms. Apparently, a number of pathological causes may cause and promote dysfunctions of PA endothelial cells and PASMCs via numerous distinctive signaling mechanisms, thereby leading to the initiation and development of PH, right ventricular failure (RVF) and even death [21,33][21][28]. The pathophysiology of PH involves continuous pulmonary vasoconstriction, endothelial cells (ECs) injury, vascular smooth muscle (VSM) damage and proliferation, intimal fibrosis, remodeling, and inflammation. These alterations, i.e., pulmonary vascular remodeling (PVR), promote an augmented pulmonary vascular resistance (by the occlusion of blood vessels), increasing the right ventricle afterload and leading to right ventricular hypertrophy and failure, and eventually death [38,39][33][34]. The enhanced pulmonary vascular reactivity is also associated with malfunctioning of endothelial cells, leading to an imbalance in the production of nitric oxide (NO), prostaglandin (PG)-I2 (also named prostacyclin), and endothelin-1. More specifically, it has been shown that patients suffering from PAH display reduced expression of endothelial nitric oxide synthase (eNOS) and NO levels in the lungs [40,41][35][36]. In addition, the expression of prostacyclin synthase is diminished in patients with severe PH [42][37]. Importantly, PGI2 triggers the synthesis of cyclic adenosine monophosphate (cAMP) and stimulates the peroxisome proliferator activated receptor-γ (PPARγ), leading to an antiproliferative effect in VSM cells (VSMCs) [43][38]. Eventually, this endothelial perturbance results in a diminished endothelium dependent pulmonary vasculature relaxation [44,45][39][40].Table 1
. WHO classification of pulmonary hypertension (PH).
|
WHO group |
Clinical classification |
Covering subtypes |
|
I |
Pulmonary arterial hypertension (PAH) |
Idiopathic; Drug and toxin-Induced; Heritable; Associated with connective tissue diseases, HIV infection, portal hypertension, schistosomiasis; PAH responder to Ca2+ channel blockers; Associated with pulmonary venous/capillaries occlusion; Persistent pulmonary hypertension of the newborn. |
|
II |
PH due to left heart diseases |
Heart failure; Valvular heart disease; Congenital or acquired cardiomyopathies; Failure with preserved/reduced ejection fraction. |
|
III |
PH due to lung disease or hypoxia |
COPD/hypoxia that includes COPD; Restrictive lung disease; Pulmonary disease with obstructive and restrictive pattern; Interstitial lung disease; Hypoxia without other lung diseases. |
|
VI |
PH due to the obstruction of pulmonary artery |
Chronic thromboembolic pulmonary hypertension (CTEPH); Other pulmonary artery obstructions. |
|
V |
PH due to unclear/multifactorial mechanisms |
Hematologic disorders; Metabolic disorders; Others. |
3. Inflammation in Pulmonary Hypertension
The inflammatory response is implicated in the development of PH, particularly in PAH subtype due to diverse molecular pathologies [59]. Perivascular inflammation precedes pulmonary vascular lesions. Endothelial injury favors the participation of chemokines involved in the recruitment of inflammatory cells and intravascular infiltration. Cytokines released from recruited inflammatory cells mediate communication between endothelial and other vascular cells, e.g., VSMCs. In PH, increased levels of IL-1, IL-6 and tumor necrosis factor (TNF)-α are exhibited [74,75,76][54][55][56]. It has been suggested that the heightened levels of IL-1 and IL-6 derives primarily from lung microvascular endothelium [77][57]. In a PH rat model induced with MCT, excessive amounts of IL-1 were found in the lungs [78][58]. Furthermore, elevated levels of IL-6 have been related with poor survival in patients with PH [79][59]. All these inflammatory alterations result in vascular remodeling, leading to increased pulmonary vascular pressures and resistance. In this context, mitochondria undergo physiological and structural changes during PH [80][60], and correspondingly ROS levels are altered [81,82][61][62]. Nuclear Factor κB (NF-κB) is a master regulator of inflammation implicated in the development of PH. NF-κB is a family of inducible transcription factors considered as the main controllers of innate immunity [83][63]. Five structurally related family subunits have been identified: p50, p51, RelA (p65), RelB, and c-Rel. The activation of these family members depends on the degradation of the inhibitor of NF-κB proteins (IκBs), which hold inactive NF-κB dimers in the cytosol upon the stimulation of determined cells. Regarding the role of NF-κB in PH, Sawada et al. in 2007 reported that the stimulation of NF-κB, leading to the activation of the vascular cell adhesion molecule (VCAM)-1, is related to the development of MCT-induced PH in rats. Moreover, the use of the NF-κB inhibitor, pyrrolidine dithiocarbamate (PDTC), decreases PH symptoms [88]. Later, Huang et al. using the same animal model demonstrated that PDTC restores endothelial cell membrane integrity by rescuing caveolin-1, leading to PH [89]. Likewise, the inhibition of NF-κB with PDTC has proven to be effective in decreasing arterial lumen obliteration in SU5416-induced PH [90]. Interestingly, the role of NF-κB in a hypoxia-induced PH model has been investigated as well. It is well known that chronic hypoxia can lead to apoptosis, vascular remodeling and ultimately PH [93]. Hypoxia-inducible factor (HIF)-1α augments its transcriptional activity in response to oxygen decline in the lung [94]. In this context, Luo et al. demonstrated that NF-κB mediates the transcriptional program of HIF-1α promoting vascular remodeling in a PH model [95]. Eventually, it has been shown that the abnormal activity and regulation of NF-κB exacerbate the inflammatory and Ca2+ responses in PASMCs from PAH patients [96].4. Mitochondria in Vascular Remodeling during PH
The mitochondria of the vascular smooth muscle cells (VSMCs) and PAECs, as in any other cells, are responsible for the synthesis of ATP, the key energetic molecule, thus a strict control of metabolism is exerted by these double-membrane-bound organelles [97][64]. In addition, mitochondria take an important place in the production and regulation of ROS, Ca2+ signaling, metabolism of glucose and FA, and apoptosis. These mechanisms are essential players in the development of PVR seen in PH disease [49][44]. During PH, a disfunction in mitochondria’s metabolism occurs, particularly a shift in energy production from OXPHOS to glycolysis and lactic acid fermentation in order to maintain ATP production and cell survival [98][65]. This phenomenon, known as the Warburg effect, was described in 1956 by Otto Warburg in tumor cells under normal oxygen conditions to support the uncontrolled growth of neoplastic tissue [99,100][66][67]. Hyperproliferation, survival and metabolic reprogramming of PASMCs and PAECs set the basis for the pathophysiology of PH [101,102][68][69]. Moreover, mitochondria in PASMCs sense the oxygen levels, and patients suffering from PH display abnormalities in this mechanism [103][70]. Furthermore, PVR involving structural changes in intima, media and adventitia is linked to a marked inflammatory process in pulmonary hypertension [49,104][44][71]. However, the precise mechanisms underlying this relationship are still uncertain and mitochondrial dysfunction may serve as an explanation. Alterations in mitochondrial respiration can lead to variations in mitochondrial membrane potential (MMP) [105][72]. MMP has shown to be either hyperpolarized [106,107,108][73][74][75] or depolarized [109][76] in PH models. Mitochondrial uncoupling proteins (UCPs) participate in the control and regulation of MMP and ROS production. Five UCP homologues have been characterized in mammals (UCP1-UCP5) [110][77]. In this regard, Pak et al. showed that the genetic ablation of UCP2 promotes the proliferation of PASMCs in mice. Additionally, they found that MMP and ROS production are increased in PASMCs from patients and in animal models of MCT- and hypoxia-induced PH [80][60]. Recently, it has been shown that heat-shock protein 90 (HSP90), in response to stress, accumulates in the mitochondria of PASMCs from PH patients to protect mitochondrial DNA (mitDNA) and preserve mitochondrial functions leading to cell survival. Moreover, the inhibition of mitochondrial HSP90 (mtHSP90) diminishes mitDNA content, restores mitochondrial bioenergetics and limits the hyperproliferative state of PASMCs [111][78]. As well as mitochondrial dysfunction, endoplasmic reticulum (ER) stress is implicated in PH pathophysiology, and an interesting interplay between mitochondrial and ER stress drives some aspects of this disease. For instance, the loss of function of bone morphogenetic protein receptor type II (BMPRII) has been shown to induce ER stress, being a critical genetic factor predisposing to PAH [112][79]. Restoring BMPRII performance and the abolishment of ER stress have been remarkable suggested as a potential treatment against PH [113,114][80][81]. Mitochondrial fragmentation complemented with ER stress have been observed in PASMCs from hypoxic-induced PH rats [115][82]. Moreover, the same work exhibits that mitochondrial fragmentation promotes ER stress through a ROS dependent mechanism and the abolishment of ER stress improves PASMCs function under hypoxic condition. More interestingly, the mitochondrial division inhibitor (Mdvi-1) decreases mitochondrial fragmentation, ER stress and improves PASMC performance [115][82]. It is known that ER stress can cause unfolded proteins to accumulate in the ER and then activate the unfolded protein response (UPR). The persistent UPR conduces to the dysfunction of mitochondria accompanied by the disturbance of mitochondria-associated ER membrane (MAM), ultimately leading to cell apoptosis [116,117][83][84]. In this context, it has been observed that S-nitroso-L-cysteine (CSNO), a derivative of NO, improves ER stress and regulates the expression of contractile smooth muscle proteins in the lungs of MCT-induced PH rats. CSNO also leads to smooth muscle relaxation via anti-inflammatory pathways, ameliorating PVR [22]. Furthermore, the disruption of MAMs diminishes mitochondrial aberrations in ECs under hypoxic stimulus through an augment in NO release and the inhibition of the proinflammatory profile induced by hypoxia [118][85].5. Mitochondrial ROS in Pulmonary Vasoconstriction and Endothelial Dysfunction
Variations in [ROS]i in pulmonary vascular cells play a role in the pathogenesis of PH. Oxidative stress and ROS signaling in PASMCs are involved in PA vasoconstriction and remodeling and, therefore in the development and progression of PH [119][86]. It is well established that mitochondria account for the most production of ROS in PASMCs [9,120][9][87]. Mitochondria are considered an important factor in PVR due to their participation in numerous proliferative signaling pathways, such as regulating ROS production, ATP balance, apoptosis, metabolism of glucose and FA, or controlling Ca2+ homeostasis. In particular, ROS produced by mitochondrial complexes I, II and III have been suggested to play an important role in the development of PH. For instance, genetic deletion of the core subunit of mitochondrial complex I, NADH dehydrogenase (ubiquinone) iron–sulfur protein 2 (NDUFS2), was reported to decrease mitROS (H2O2) production and abolish hypoxia-induced pulmonary vasoconstriction (HPV) in mice and rats [121][88]. In this context, HPV is known to redirect blood flow from hypoxic to better ventilated areas of the lung. Moreover, HPV and arterial occlusion are important causes of PH as they decrease blood flow and increase vascular resistance. HPV appears to be mediated in part by an increase in intracellular Ca2+ and ROS signaling. [122][89].6. Mitochondrial Ca2+, ROS, and Glutaminolysis
The homeostasis of intracellular Ca2+ concentration ([Ca2+]i) is crucial to maintain the vascular tone. At rest, basal [Ca2+]i is tightly regulated to be around 100 nM. After cellular stimulation with a vasoconstrictor agonist, such as norepinephrine, endothelin, vasopressin, etc., [Ca2+]i increases reaching values between 500 nM and 1 mM [128][90]. ROS facilitates the dissociation of FKBP12.6 from ryanodine receptor 2 (RyR2) to activate the channel [125,129,130,131][91][92][93][94]. Moreover, weit haves demonstrated that Rieske iron–sulfur protein (RISP) knockdown (KD) abolishes the hypoxic ROS formation in isolated PASMCs, whereas RISP overexpression produces the opposite effect. RISP KD also inhibits the hypoxic increase in [Ca2+]i in PASMCs [124,132][95][96]. Most recently, weit showed that the dissociation of the FKBP12.6/RyR2 complex (induced by chronic hypoxia) causes sarcoplasmic reticulum (SR) Ca2+ leak and increases [Ca2+]i in PASMCs (Figure 1), thereby leading to subsequent pulmonary artery remodeling and vasoconstriction. These events may occur due to the mitochondrial RISP-dependent ROS generation and the subsequent RyR2 oxidation [5,9][5][9]. Furthermore, FKBP12.6 is also specifically bound to big-conductance Ca2+-activated K+ (BKCa) channels in VSMCs. These channels as well as RyRs are essential proteins in mediating vascular smooth muscle tone. Mitochondrial Ca2+ uniporter (MCU) regulates mitochondrial Ca2+ (mitCa2+) allowing Ca2+ uptake. WeIt have reported that Ca2+ release mediated by hypoxic or RyR simulation evokes an improved performance in the activity of MCU. The increased MCU leads to the generation of mitROS dependent on ROS, provoking a positive feedback mechanism to potentiate hypoxia-initiated mitROS in PASMCs [9]. This finding indicates an important role of mitROS and MCU in HPV and associated PH. Alterations in ROS production can alter the physiology of ion channels in PASMCs and induce a large increase in [Ca2+]i [133][97].Glutaminolysis is a mitochondrial process responsible for obtaining cellular energy from the deamination of glutamine to glutamate by glutaminase (GLS1) [139]. Subsequently, glutamate is converted to α-ketoglutarate (α-KG) by glutamate dehydrogenase. This process (anaplerotic reactions) helps to replenish the intermediates of the TCA cycle after they have been consumed and provides energy especially for proliferating cells. The increase in glutaminolysis leads to increased expression of GLS1 and increased uptake of glutamine by the pulmonary vasculature, resulting in increased glutamate production by pulmonary vascular cells and promoting PH. Activation of the former transcriptional coactivators triggers upregulation of GLS1 and leads to glutaminolysis, which maintains the hyperproliferative state and migration of pulmonary vascular cells in PH (Figure 2) [141].
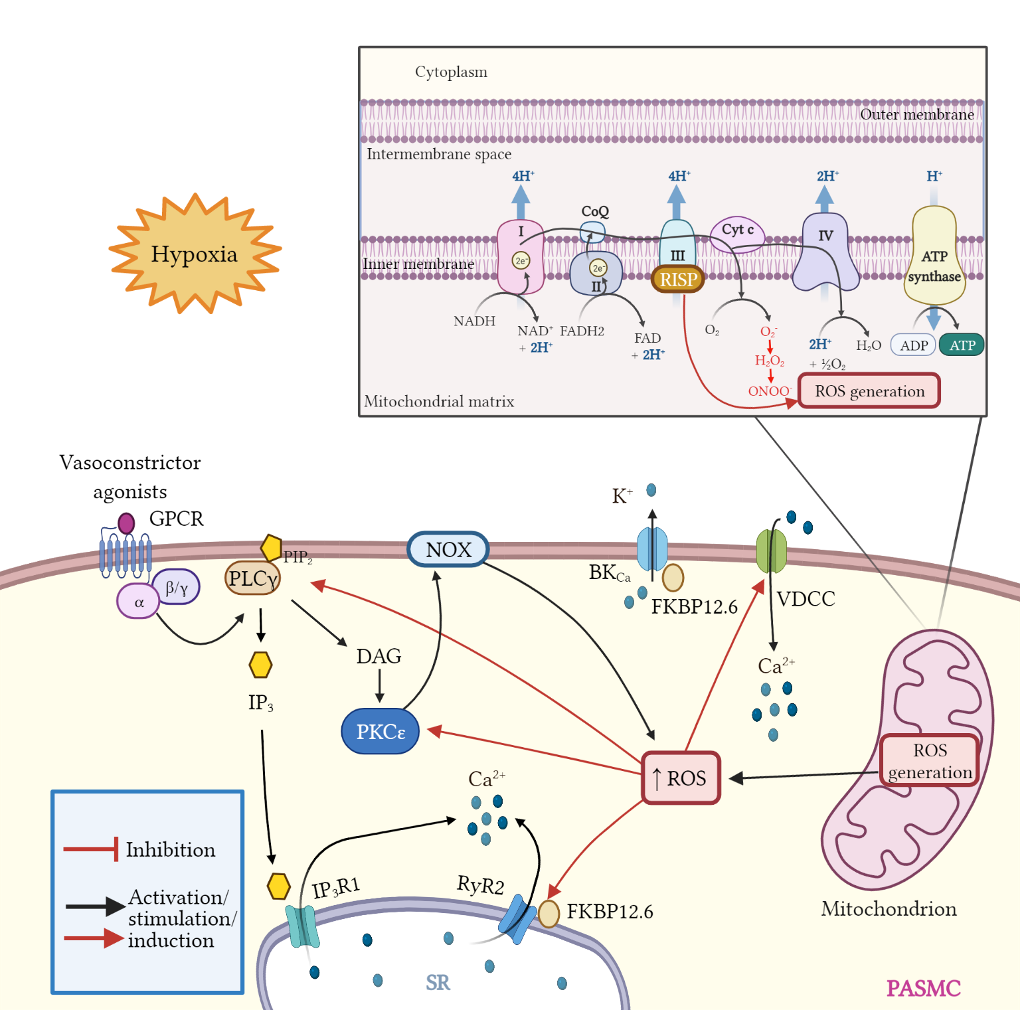 Figure 1. Schematic diagram of mitROS generation and signaling effects; crosstalk between ROS and Ca2+ signaling in PASMCs. Mitochondria are the main source of ROS in pulmonary artery smooth muscle cells (PASMCs). During ATP synthesis in the electron transport chain (ETC), the coupling between the proton gradient on either side of the inner mitochondrial membrane leads to the production of ROS. Briefly, electrons are transferred from nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) to molecular oxygen. In the process, protons are pumped from the mitochondrial matrix to the intermembrane space, and oxygen is reduced to form water. Hypoxia enhances mitROS production, contributing to the increase in [Ca2+]i and to hypoxia-induced pulmonary vasoconstriction. Rieske iron-sulfur protein (RISP), a catalytic subunit of the complex III of the mitochondrial ETC serves as a primary molecule in intracellular ROS generation in PASMCs, especially under hypoxic conditions. Moreover, mitROS and vasoconstrictor agonists via GPCR activation stimulate the PLCγ and PKCε signaling pathways. PLCγ induces the formation of IP3 and DAG, evoking the opening of IP3R1 and Ca2+ release from sarcoplasmic reticulum (SR). Also, mitROS augment the activity of PKCε which in turn stimulates NOX and propagates ROS generation in a process named ROS-induced ROS generation (RIRG). Moreover, ROS enable the dissociation of FK506 binding protein 12.6 (FKBP12.6) from ryanodine receptor 2 (RyR2) favoring the opening of this channel and enhancing Ca2+ release. In addition, FKBP12.6 is physically bound to large conductance K+ channels (BKCa) regulating its open probability. Finally, ROS upregulate voltage dependent Ca2+ channels (VDCCs) which further contribute to increasing [Ca2+]i, leading to persistent vasoconstriction observed in PH.
Figure 1. Schematic diagram of mitROS generation and signaling effects; crosstalk between ROS and Ca2+ signaling in PASMCs. Mitochondria are the main source of ROS in pulmonary artery smooth muscle cells (PASMCs). During ATP synthesis in the electron transport chain (ETC), the coupling between the proton gradient on either side of the inner mitochondrial membrane leads to the production of ROS. Briefly, electrons are transferred from nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) to molecular oxygen. In the process, protons are pumped from the mitochondrial matrix to the intermembrane space, and oxygen is reduced to form water. Hypoxia enhances mitROS production, contributing to the increase in [Ca2+]i and to hypoxia-induced pulmonary vasoconstriction. Rieske iron-sulfur protein (RISP), a catalytic subunit of the complex III of the mitochondrial ETC serves as a primary molecule in intracellular ROS generation in PASMCs, especially under hypoxic conditions. Moreover, mitROS and vasoconstrictor agonists via GPCR activation stimulate the PLCγ and PKCε signaling pathways. PLCγ induces the formation of IP3 and DAG, evoking the opening of IP3R1 and Ca2+ release from sarcoplasmic reticulum (SR). Also, mitROS augment the activity of PKCε which in turn stimulates NOX and propagates ROS generation in a process named ROS-induced ROS generation (RIRG). Moreover, ROS enable the dissociation of FK506 binding protein 12.6 (FKBP12.6) from ryanodine receptor 2 (RyR2) favoring the opening of this channel and enhancing Ca2+ release. In addition, FKBP12.6 is physically bound to large conductance K+ channels (BKCa) regulating its open probability. Finally, ROS upregulate voltage dependent Ca2+ channels (VDCCs) which further contribute to increasing [Ca2+]i, leading to persistent vasoconstriction observed in PH.
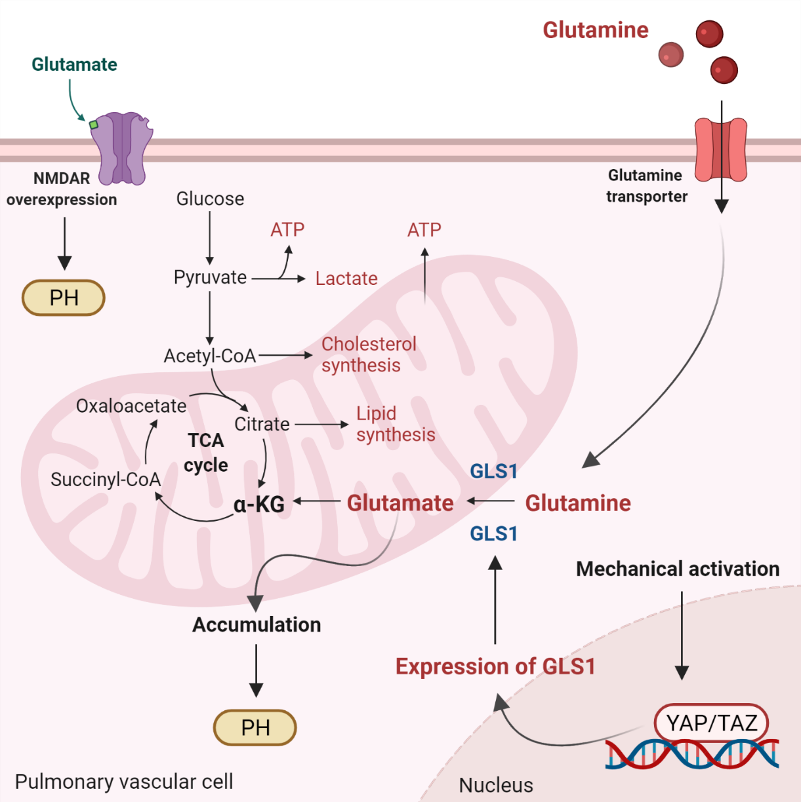 Figure 2. Glutaminolysis and glutamate accumulation contribute to pulmonary hypertension (PH). Glutaminolysis is a mitochondrial process responsible for obtaining cellular energy from the breakdown of glutamine. In this cellular pathway, glutamine is converted into glutamate, aspartate, CO2, pyruvate, lactate, alanine, and citrate. Initially, glutamine enters the pulmonary vascular cells via a glutamine transporter and is deaminated to glutamate by glutaminase (GLS1). Subsequently, glutamate is converted to a-ketoglutarate (a-KG) by glutamate dehydrogenase. a kg enters the tricarboxylic acid (TCA) cycle, where it is decarboxylated by a kg dehydrogenase to succinyl-CoA and CO2, providing energy for proliferating cells. Accumulation of glutamate in pulmonary vascular cells promotes PH. In addition, stiffening of the extracellular matrix in remodeled pulmonary cells activates the transcriptional coactivators Yes-associated protein 1 (YAP) and TAZ, leading to upregulation of GLS1 and enhanced glutaminolysis. Furthermore, in remodeled pulmonary arteries, the N-methyl-d-aspartate receptor (NMDAR) is overexpressed and overactivated.
Figure 2. Glutaminolysis and glutamate accumulation contribute to pulmonary hypertension (PH). Glutaminolysis is a mitochondrial process responsible for obtaining cellular energy from the breakdown of glutamine. In this cellular pathway, glutamine is converted into glutamate, aspartate, CO2, pyruvate, lactate, alanine, and citrate. Initially, glutamine enters the pulmonary vascular cells via a glutamine transporter and is deaminated to glutamate by glutaminase (GLS1). Subsequently, glutamate is converted to a-ketoglutarate (a-KG) by glutamate dehydrogenase. a kg enters the tricarboxylic acid (TCA) cycle, where it is decarboxylated by a kg dehydrogenase to succinyl-CoA and CO2, providing energy for proliferating cells. Accumulation of glutamate in pulmonary vascular cells promotes PH. In addition, stiffening of the extracellular matrix in remodeled pulmonary cells activates the transcriptional coactivators Yes-associated protein 1 (YAP) and TAZ, leading to upregulation of GLS1 and enhanced glutaminolysis. Furthermore, in remodeled pulmonary arteries, the N-methyl-d-aspartate receptor (NMDAR) is overexpressed and overactivated.
7. Ketones and Mitochondrial Signaling
Ketone bodies, or simply ketones, are highly polar molecules produced by β-oxidation of fatty acids in the mitochondria of the liver cells. However, these molecules may be produced by enterocytes, astrocytes, and kidney ECs to a lesser extent [24,26,143][98][99][100]. Ketones are produced in response to reduced glucose availability, e.g., during periods of prolonged fasting, high-performance exercise, or a pathophysiological state, such as type I diabetes [144,145][101][102]. It has been postulated that patients with PAH have reduced oral glucose tolerance and lipid and ketone metabolism predominate over the glucose control [58][53]. Ketone’s metabolism is divided in ketogenesis and ketolysis. Ketogenesis, which mainly occurs in perivenous hepatocytes, produces three molecules: acetone, acetoacetate, and β-HB [146][103], which represents the most abundant ketone body [147,148,149][104][105][106]. It is well known that adipocytes store great amounts of energy as fatty acids [150][107]. When fasting or exercising, glycogen stores are used in the beginning. Once glycogen is depleted, fatty acids from adipocytes are transferred into the liver by the enzyme carnitine palmitoyltransferase (CPT-1), where they are metabolized in the mitochondria to form ketone bodies [151,152][108][109]. These lipid derivatives enter the systemic circulation and reach highly metabolic tissues, e.g., muscles and nervous system, which convert ketones into acetyl coenzyme A (acetyl-CoA) for alternative energy metabolism. The detailed process occurs as follows: two molecules of acetyl-CoA are biotransformed in acetoacetyl-CoA by the action of the acetyl-CoA acetyltransferase (ACAT), a thiolase [153][110]. Then, 3-hydroxy-3-methylglutaryl Co-A synthase (HMGS2) condenses acetyl-CoA with acetoacetyl-CoA to produce HMG-CoA. Afterward, HMG-CoA is broken into acetoacetate via HMG-CoA lyase (HMGCL). Finally, acetoacetate is further bioconverted to acetone (by decarboxylation) or to β-HB by the action of 3-hydroxybutyrate dehydrogenase (BDH1) [148][105]. On the other hand, throughout ketolysis, acetoacetate and β-HB are used as an energy source by the mitochondria of several extrahepatic tissues [148,154][105][111]. β-HB is transformed to acetoacetate by the BDH1, and this last product is turned into acetoacetyl-CoA by the enzyme beta-ketoacyl-CoA transferase (OXCT1 or SCOT). Acetoacetyl-CoA is separated by thiolase in two molecules of acetyl-CoA [155][112], that enters the TCA, and subsequently the oxidative phosphorylation, resulting in the generation of ATP [148,156][105][113]. Importantly, acetone (the other ketone body) cannot be biotransformed into acetyl-CoA and ends up eliminated through urine or exhaled [157,158][114][115]. High glucose levels may elicit the over production of ROS, mainly through NOX4 activity in endothelial cells [161][116]. In addition, in these cells, hyperglycemia triggers NF-κB signaling pathway leading to the upregulation of proinflammatory cytokines and endothelial adhesion molecules [162][117]. The induction of endothelial selectin (E-selectin), intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and increased mononuclear-endothelial adhesion in addition to ROS generation and NF-κB activity increase vascular permeability and facilitates endothelial barrier dysfunction [162][117]. The role of ketones in the development of PH remains elusive. In this regard, the evidence indicates a relationship between the RVF occurring in PH and metabolic disorders (risk factors/comorbidities in human PH), such as high blood sugar [58][53], insulin resistance [164][118], dyslipidemia [165][119], and abdominal obesity [166][120]. It is well known that 70–90% of ATP produced in heart comes from the oxidation of FA in mitochondria. The residual percentage is generated by the metabolism of glucose, ketone bodies and amino acids [171][121]. In this context, Peroxisome proliferator-activated receptor γ (PPARγ) has been shown to regulate glucose and FA metabolism in adipocytes, hepatocytes, skeletal muscle, and pancreatic β cells [172,173,174][122][123][124]. PPARs belong to the superfamily of nuclear receptors serving as ligand-activated transcription factors. This receptor subfamily is composed of three members, PPARα, PPARγ and PPARδ, which combine with retinoid X receptors (RXRs) forming heterodimers and binding to specific DNA sites to promote genic transcription [173][123]. PPARγ is a master regulator of adipogenesis expressed mostly in adipose tissue and liver, as well as PPARα. PPARδ is ubiquitously expressed and all three subtypes are expressed in heart [175][125]. Emerging evidence highlights that PPARγ acts as a strong, protective regulator in PAH [176][126], PASMCs [177][127], and PAECs [178][128]. As it concerns, Legchenko et al., in a SU5416/hypoxia-induced PAH rat model, demonstrated that the oral administration of pioglitazone (a PPARγ agonist) fully abolishes severe PAH and vascular remodeling, and prevents RVF (Figure 3).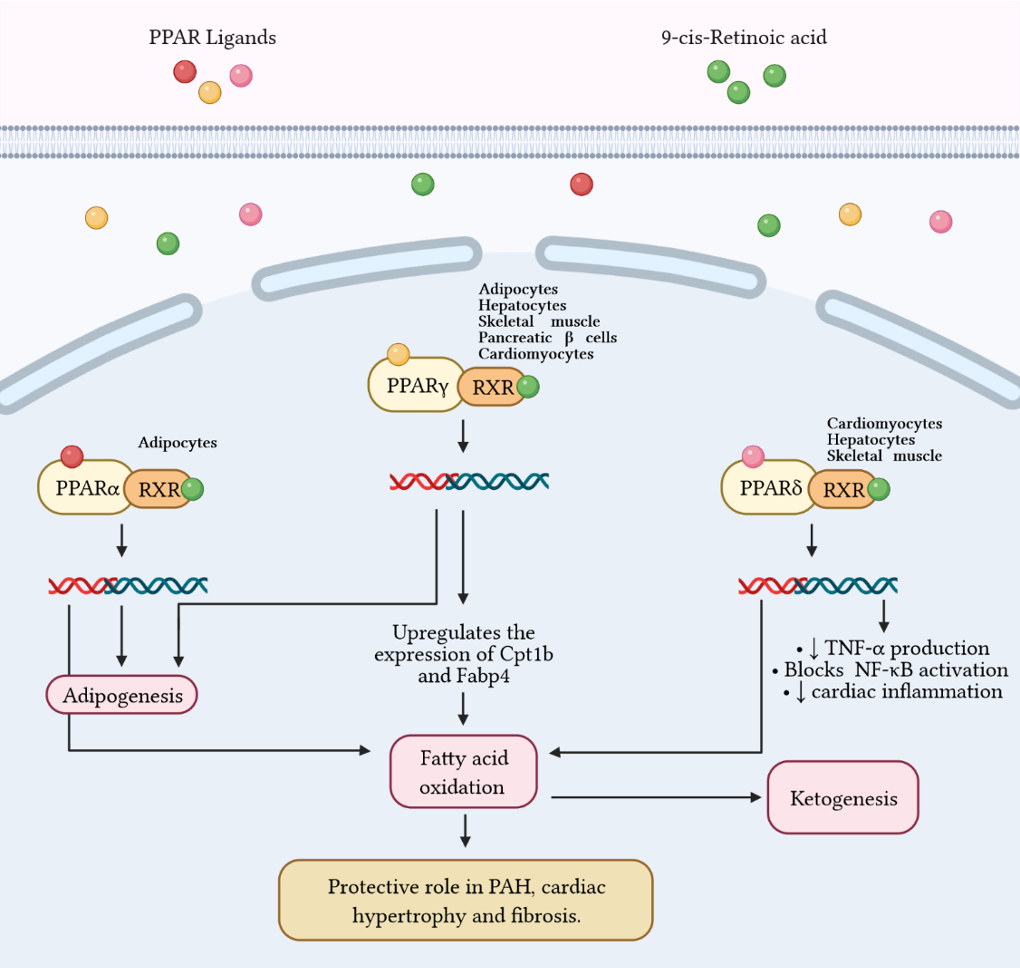 Figure 3 Fatty acid metabolism and its byproducts (ketones) play an important role in the development of right ventricular failure (RVF) and pulmonary arterial hypertension (PAH). Peroxisome proliferator-activated receptors (PPAR) belong to the superfamily of nuclear receptors that serve as ligand-activated transcription factors and consist of three members, PPARa, PPARg, and PPARd. These receptors together with retinoid X receptors (RXR) form heterodimers and bind to specific DNA sites to promote genetic transcription. PPARa is a master regulator of adipogenesis expressed mainly in adipose tissue and liver, as is PPARg. Additionally, PPARg regulates glucose and fatty acid metabolism in adipocytes, hepatocytes, skeletal muscle, and pancreatic b-cells. PPARg agonists, such as pioglitazone, increase the expression of Cpt1b and Fabp4, proteins involved in fatty acid oxidation and transport in cardiomyocytes. These effects favor mitochondrial fatty acid oxidation and ATP production, leading to reversal of cardiac hypertrophy, fibrosis, and eliminating severe PAH. Furthermore, PPARd stimulates fatty acid oxidation, decreases right ventricle hypertrophy and pulmonary congestion. In cardiac inflammation, PPARd blocks nuclear factor kB (NF-kB) activation and inhibits tumor necrosis factor (TNF)-a synthesis.
Figure 3 Fatty acid metabolism and its byproducts (ketones) play an important role in the development of right ventricular failure (RVF) and pulmonary arterial hypertension (PAH). Peroxisome proliferator-activated receptors (PPAR) belong to the superfamily of nuclear receptors that serve as ligand-activated transcription factors and consist of three members, PPARa, PPARg, and PPARd. These receptors together with retinoid X receptors (RXR) form heterodimers and bind to specific DNA sites to promote genetic transcription. PPARa is a master regulator of adipogenesis expressed mainly in adipose tissue and liver, as is PPARg. Additionally, PPARg regulates glucose and fatty acid metabolism in adipocytes, hepatocytes, skeletal muscle, and pancreatic b-cells. PPARg agonists, such as pioglitazone, increase the expression of Cpt1b and Fabp4, proteins involved in fatty acid oxidation and transport in cardiomyocytes. These effects favor mitochondrial fatty acid oxidation and ATP production, leading to reversal of cardiac hypertrophy, fibrosis, and eliminating severe PAH. Furthermore, PPARd stimulates fatty acid oxidation, decreases right ventricle hypertrophy and pulmonary congestion. In cardiac inflammation, PPARd blocks nuclear factor kB (NF-kB) activation and inhibits tumor necrosis factor (TNF)-a synthesis.
8. Conclusions
Mitochondria are involved in essential cellular regulatory and homeostatic process in the cardiovascular system and particularly in VSMCs. Mitochondrial dysfunction has been widely associated with several diseases including PH. These organelles exert a strict control of ROS and ketones production. Uncontrolled ROS generation and its crosstalk with Ca2+ signaling have been shown to contribute and aggravate pulmonary hypertension. Thus, anti-ROS therapies targeting implicated proteins such as RISP should be investigated as novel alternatives for the treatment of this ailment. On the other hand, ketone bodies seem to offer a protection against oxidative stress damage in vitro and in vivo; however, they also may exert anti-inflammatory or pro-inflammatory roles in the cardiovascular system, and further research is needed to understand their different roles. Collectively, a better comprehension of the unique roles of RISP-dependent mitochondrial ROS and their specific interactions with mitochondrial Ca2+ signaling, NF-κB-mediated inflammatory responses, and ketone-associated oxidative stress can significantly improve ourthe understanding of the molecular pathogenesis of PH and associated RVF. This new knowledge may also help to develop and implement innovative therapies in the treatment of PH and other vascular diseases.References
- Bruno, S.R.; Anathy, V. Lung epithelial endoplasmic reticulum and mitochondrial 3D ultrastructure: A new frontier in lung diseases. Histochem. Cell Biol. 2021, 155, 291–300.
- Bertram, R.; Pedersen, M.G.; Luciani, D.S.; Sherman, A. A simplified model for mitochondrial ATP production. J. Theor. Biol. 2006, 243, 575–586.
- Grumbach, I.M.; Nguyen, E.K. Metabolic Stress. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 991–997.
- Waypa, G.B.; Marks, J.D.; Guzy, R.D.; Mungai, P.T.; Schriewer, J.M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am. J. Respir. Crit. Care Med. 2013, 187, 424–432.
- Mei, L.; Zheng, Y.-M.; Song, T.; Yadav, V.R.; Joseph, L.C.; Truong, L.; Kandhi, S.; Barroso, M.M.; Takeshima, H.; Judson, M.A.; et al. Rieske iron-sulfur protein induces FKBP12.6/RyR2 complex remodeling and subsequent pulmonary hypertension through NF-κB/cyclin D1 pathway. Nat. Commun. 2020, 11, 3527.
- Rathore, R.; Zheng, Y.-M.; Niu, C.-F.; Liu, Q.-H.; Korde, A.; Ho, Y.-S.; Wang, Y.-X. Hypoxia activates NADPH oxidase to increase i and i through the mitochondrial ROS-PKCɛ signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 1223–1231.
- Truong, L.; Zheng, Y.-M.; Wang, Y.-X. Mitochondrial Rieske iron–sulfur protein in pulmonary artery smooth muscle: A key primary signaling molecule in pulmonary hypertension. Arch. Biochem. Biophys. 2020, 683, 108234.
- Yadav, V.R.; Song, T.; Mei, L.; Joseph, L.; Zheng, Y.-M.; Wang, Y.-X. PLCγ1-PKCε-IP3R1 signaling plays an important role in hypoxia-induced calcium response in pulmonary artery smooth muscle cells. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2018, 314, L724–L735.
- Yang, Z.; Song, T.; Truong, L.; Reyes-Garcia, J.; Wang, L.; Zheng, Y.-M.; Wang, Y.-X. Important role of sarcoplasmic reticulum Ca2+ release via ryanodine receptor-2 channel in hypoxia-induced rieske iron–sulfur protein-mediated mitochondrial reactive oxygen species generation in pulmonary artery smooth muscle cells. Antioxid. Redox Signal. 2020, 32, 447–462.
- Mohamed, R.; Dayati, P.; Mehr, R.N.; Kamato, D.; Seif, F.; Babaahmadi-Rezaei, H.; Little, P.J. Transforming growth factor–β1 mediated CHST11 and CHSY1 mRNA expression is ROS dependent in vascular smooth muscle cells. J. Cell Commun. Signal. 2019, 13, 225–233.
- Salazar, G.; Huang, J.; Feresin, R.; Zhao, Y.; Griendling, K. Zinc regulates Nox1 expression through a NF-κB and mitochondrial ROS dependent mechanism to induce senescence of vascular smooth muscle cells. Free Radic. Biol. Med. 2017, 108, 225–235.
- Pieczenik, S.R.; Neustadt, J. Mitochondrial dysfunction and molecular pathways of disease. Exp. Mol. Pathol. 2007, 83, 84–92.
- Chen, J.; Stimpson, S.; Fernandez-Bueno, G.A.; Mathews, C.E. Mitochondrial reactive oxygen species and type 1 diabetes. Antioxid. Redox Signal. 2018, 29, 1361–1372.
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285.
- Favero, G.; Bonomini, F.; Franco, C.; Rezzani, R. Mitochondrial dysfunction in skeletal muscle of a fibromyalgia model: The potential benefits of melatonin. Int. J. Mol. Sci. 2019, 20, 765.
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13.
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184.
- Cano Sanchez, M.; Lancel, S.; Boulanger, E.; Neviere, R. Targeting oxidative stress and mitochondrial dysfunction in the treatment of impaired wound healing: A systematic review. Antioxidants 2018, 7, 98.
- Zhu, Y.; Li, M.; Lu, Y.; Li, J.; Ke, Y.; Yang, J. Ilexgenin A inhibits mitochondrial fission and promote Drp1 degradation by Nrf2-induced PSMB5 in endothelial cells. Drug Dev. Res. 2019, 80, 481–489.
- Chen, J.; Wang, Y.; Dong, M.; Zhang, B.; Luo, Y.; Niu, W.; Li, Z. Reoxygenation reverses hypoxic pulmonary arterial remodeling by inducing smooth muscle cell apoptosis via reactive oxygen species–mediated mitochondrial dysfunction. J. Am. Heart Assoc. 2017, 6, e005602.
- Huetsch, J.C.; Suresh, K.; Shimoda, L.A. Regulation of smooth muscle cell proliferation by NADPH oxidases in pulmonary hypertension. Antioxidants 2019, 8, 56.
- Wang, M.; Luo, P.; Shi, W.; Guo, J.; Huo, S.; Yan, D.; Peng, L.; Zhang, C.; Lv, J.; Lin, L.; et al. S-nitroso-L-cysteine ameliorated pulmonary hypertension in the MCT-induced rats through anti-ROS and anti-inflammatory pathways. Oxidtive Med. Cell. Longev. 2021, 2021, 6621232.
- Zhuan, B.; Yu, Y.; Yang, Z.; Zhao, X.; Li, P. Mechanisms of oxidative stress effects of the NADPH oxidase-ROS-NF-kappaB transduction pathway and VPO1 on patients with chronic obstructive pulmonary disease combined with pulmonary hypertension. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3459–3464.
- Escribano-Subias, P.; Blanco, I.; Meseguer, M.L.; Lopez-Guarch, C.J.; Roman, A.; Morales, P.; Castillo-Palma, M.J.; Segovia, J.; Gómez-Sanchez, M.A.; Barberà, J.A. Survival in pulmonary hypertension in Spain: Insights from the Spanish registry. Eur. Respir. J. 2012, 40, 596–603.
- Ling, Y.; Johnson, M.K.; Kiely, D.G.; Condliffe, R.; Elliot, C.A.; Gibbs, J.S.R.; Howard, L.; Pepke-Zaba, J.; Sheares, K.K.K.; Corris, P.A.; et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: Results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am. J. Respir. Crit. Care Med. 2012, 186, 790–796.
- Peacock, A.J.; Murphy, N.F.; McMurray, J.J.V.; Caballero, L.; Stewart, S. An epidemiological study of pulmonary arterial hypertension. Eur. Respir. J. 2007, 30, 104–109.
- Wijeratne, D.T.; Lajkosz, K.; Brogly, S.B.; Lougheed, M.D.; Jiang, L.; Housin, A.; Barber, D.; Johnson, A.; Doliszny, K.M.; Archer, S.L. Increasing incidence and prevalence of World Health Organization groups 1 to 4 pulmonary hypertension: A population-based cohort study in Ontario, Canada. Circ. Cardiovasc. Qual. Outcomes 2018, 11, e003973.
- Prasad, K. AGE–RAGE Stress in the pathophysiology of pulmonary hypertension and its treatment. Int. J. Angiol. 2019, 28, 71–79.
- Hoeper, M.M.; Huscher, D.; Ghofrani, H.A.; Delcroix, M.; Distler, O.; Schweiger, C.; Grunig, E.; Staehler, G.; Rosenkranz, S.; Halank, M.; et al. Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: Results from the COMPERA registry. Int. J. Cardiol. 2013, 168, 871–880.
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.-F.; Chabot, F.; et al. Pulmonary arterial hypertension in France: Results from a national registry. Am. J. Respir. Crit. Care Med. 2006, 173, 1023–1030.
- Benza, R.L.; Gomberg-Maitland, M.; Miller, D.P.; Frost, A.; Frantz, R.P.; Foreman, A.J.; Badesch, D.B.; McGoon, M.D. The REVEAL registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest 2012, 141, 354–362.
- Jacobs, W.; van de Veerdonk, M.C.; Trip, P.; Man, F.H.-D.; Heymans, M.W.; Marcus, J.T.; Kawut, S.M.; Bogaard, H.-J.; Boonstra, A.; Noordegraaf, A.V. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest 2014, 145, 1230–1236.
- Sakuma, M.; Toyoda, S.; Inoue, T.; Node, K. Inflammation in pulmonary artery hypertension. Vasc. Pharmacol. 2019, 118–119, 106562.
- Singh, I.; Oliveira, R.; Naeije, R.; Rahaghi, F.N.; Oldham, W.M.; Systrom, D.M.; Waxman, A.B. Pulmonary vascular distensibility and early pulmonary vascular remodeling in pulmonary hypertension. Chest 2019, 156, 724–732.
- Giaid, A.; Saleh, D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1995, 333, 214–221.
- Kaneko, F.T.; Arroliga, A.C.; Dweik, R.A.; Comhair, S.A.; Laskowski, D.; Oppedisano, R.; Thomassen, M.J.; Erzurum, S.C. Biochemical reaction products of nitric oxide as quantitative markers of primary pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1998, 158, 917–923.
- Tuder, R.M.; Cool, C.D.; Geraci, M.W.; Wang, J.; Abman, S.H.; Wright, L.; Badesch, D.; Voelkel, N.F. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1999, 159, 1925–1932.
- Falcetti, E.; Hall, S.M.; Phillips, P.G.; Patel, J.; Morrell, N.W.; Haworth, S.G.; Clapp, L.H. Smooth muscle proliferation and role of the prostacyclin (IP) receptor in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2010, 182, 1161–1170.
- Humbert, M.; Montani, D.; Perros, F.; Dorfmüller, P.; Adnot, S.; Eddahibi, S. Endothelial cell dysfunction and cross talk between endothelium and smooth muscle cells in pulmonary arterial hypertension. Vasc. Pharmacol. 2008, 49, 113–118.
- Wilson, J.L.; Warburton, R.; Taylor, L.; Toksoz, D.; Hill, N.; Polgar, P. Unraveling endothelin-1 induced hypercontractility of human pulmonary artery smooth muscle cells from patients with pulmonary arterial hypertension. PLoS ONE 2018, 13, e0195780.
- Wang, X.-Y.; Mo, D.; Tian, W.; Liu, X.-X.; Zhou, Y.-G.; Sun, Y.; Feng, Y.-D.; Xiao, X.; Hao, X.-W.; Zhang, H.-N.; et al. Inhibition of RhoA/ROCK signaling pathway ameliorates hypoxic pulmonary hypertension via HIF-1α-dependent functional TRPC channels. Toxicol. Appl. Pharmacol. 2019, 369, 60–72.
- Guo, S.; Shen, Y.; He, G.; Wang, T.; Xu, D.; Wen, F. Involvement of Ca2+-activated K+ channel 3.1 in hypoxia-induced pulmonary arterial hypertension and therapeutic effects of TRAM-34 in rats. Biosci. Rep. 2017, 37, BSR20170763.
- Lee, H.; Kim, K.C.; Hong, Y.M. Change of voltage-gated potassium channel 1.7 expressions in monocrotaline-induced pulmonary arterial hypertension rat model. Korean J. Pediatr. 2018, 61, 271–278.
- Tuder, R.M. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res. 2017, 367, 643–649.
- George, M.P.; Champion, H.C.; Simon, M.; Guyach, S.; Tarantelli, R.; Kling, H.M.; Brower, A.; Janssen, C.; Murphy, J.; Carney, J.P.; et al. Physiologic changes in a nonhuman primate model of HIV-associated pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 48, 374–381.
- Graham, B.B.; Bandeira, A.P.; Morrell, N.W.; Butrous, G.; Tuder, R.M. Schistosomiasis-associated pulmonary hypertension: Pulmonary vascular disease: The global perspective. Chest 2010, 137, 20S–29S.
- McMahan, Z.H.; Hummers, L.K. Systemic sclerosis—Challenges for clinical practice. Nat. Rev. Rheumatol. 2013, 9, 90–100.
- Li, K.; Li, Y.; Yu, Y.; Ding, J.; Huang, H.; Chu, C.; Hu, L.; Yu, Y.; Cao, Y.; Xu, P.; et al. Bmi-1 alleviates adventitial fibroblast senescence by eliminating ROS in pulmonary hypertension. BMC Pulm. Med. 2021, 21, 80.
- Li, X.; Hu, B.; Wang, L.; Xia, Q.; Ni, X. P2X7 receptor-mediated phenotype switching of pulmonary artery smooth muscle cells in hypoxia. Mol. Biol. Rep. 2021, 48, 2133–2142.
- Zhu, L.; Liu, F.; Hao, Q.; Feng, T.; Chen, Z.; Luo, S.; Xiao, R.; Sun, M.; Zhang, T.; Fan, X.; et al. Dietary geranylgeranyl pyrophosphate counteracts the benefits of statin therapy in experimental pulmonary hypertension. Circulation 2021, 143, 1775–1792.
- Bueno-Beti, C.; Hadri, L.; Hajjar, R.J.; Sassi, Y. The Sugen 5416/Hypoxia mouse model of pulmonary arterial hypertension. In Experimental Models of Cardiovascular Diseases; Humana Press: New York, NY, USA, 2018; Volume 1816, pp. 243–252.
- Morciano, G.; Vitto, V.; Bouhamida, E.; Giorgi, C.; Pinton, P. Mitochondrial bioenergetics and dynamism in the failing heart. Life 2021, 11, 436.
- Mey, J.T.; Hari, A.; Axelrod, C.L.; Fealy, C.E.; Erickson, M.L.; Kirwan, J.P.; Dweik, R.A.; Heresi, G.A. Lipids and ketones dominate metabolism at the expense of glucose control in pulmonary arterial hypertension: A hyperglycaemic clamp and metabolomics study. Eur. Respir. J. 2020, 55, 1901700.
- Groth, A.; Vrugt, B.; Brock, M.; Speich, R.; Ulrich, S.; Huber, L.C. Inflammatory cytokines in pulmonary hypertension. Respir. Res. 2014, 15, 47.
- Fujita, M.; Mason, R.J.; Cool, C.; Shannon, J.M.; Hara, N.; Fagan, K.A. Pulmonary hypertension in TNF-α-overexpressing mice is associated with decreased VEGF gene expression. J. Appl. Physiol. 2002, 93, 2162–2170.
- Humbert, M.; Monti, G.; Brenot, F.; Sitbon, O.; Portier, A.; Grangeot-Keros, L.; Duroux, P.; Galanaud, P.; Simonneau, G.; Emilie, D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1995, 151, 1628–1631.
- Barzilai, S.; Blecher-Gonen, R.; Barnett-Itzhaki, Z.; Zauberman, A.; Lebel-Haziv, Y.; Amit, I.; Alon, R. M-sec regulates polarized secretion of inflammatory endothelial chemokines and facilitates CCL2-mediated lymphocyte transendothelial migration. J. Leukoc. Biol. 2016, 99, 1045–1055.
- Voelkel, N.F.; Tuder, R.M.; Bridges, J.; Arend, W.P. Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am. J. Respir. Cell Mol. Biol. 1994, 11, 664–675.
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927.
- Pak, O.; Sommer, N.; Hoeres, T.; Bakr, A.; Waisbrod, S.; Sydykov, A.; Haag, D.; Esfandiary, A.; Kojonazarov, B.; Veit, F.; et al. Mitochondrial hyperpolarization in pulmonary vascular remodeling. Mitochondrial uncoupling protein deficiency as disease model. Am. J. Respir. Cell Mol. Biol. 2013, 49, 358–367.
- Sommer, N.; Strielkov, I.; Pak, O.; Weissmann, N. Oxygen sensing and signal transduction in hypoxic pulmonary vasoconstriction. Eur. Respir. J. 2016, 47, 288–303.
- Weir, E.K.; Archer, S.L. The role of redox changes in oxygen sensing. Respir. Physiol. Neurobiol. 2010, 174, 182–191.
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105.
- Yang, M.; Dart, C.; Kamishima, T.; Quayle, J.M. Hypoxia and metabolic inhibitors alter the intracellular ATP:ADP ratio and membrane potential in human coronary artery smooth muscle cells. PeerJ 2020, 8, e10344.
- Xu, W.; Janocha, A.J.; Erzurum, S.C. Metabolism in pulmonary hypertension. Annu. Rev. Physiol. 2021, 83, 551–576.
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270.
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314.
- Boehme, J.; Sun, X.; Tormos, K.V.; Gong, W.; Kellner, M.; Datar, S.A.; Kameny, R.J.; Yuan, J.X.-J.; Raff, G.W.; Fineman, J.R.; et al. Pulmonary artery smooth muscle cell hyperproliferation and metabolic shift triggered by pulmonary overcirculation. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H944–H957.
- Dabral, S.; Tian, X.; Kojonazarov, B.; Savai, R.; Ghofrani, H.A.; Weissmann, N.; Florio, M.; Sun, J.; Jonigk, D.; Maegel, L.; et al. Notch1 signalling regulates endothelial proliferation and apoptosis in pulmonary arterial hypertension. Eur. Respir. J. 2016, 48, 1137–1149.
- Xu, S.; Xu, X.; Zhang, J.; Ying, K.; Shao, Y.; Zhang, R. Pulmonary hypertension as a manifestation of mitochondrial disease: A case report and review of the literature. Medicine 2017, 96, e8716.
- Thistlethwaite, P.A. Linking vascular remodeling and inflammation in pulmonary arterial hypertension: Is there a common root cause? Am. J. Respir. Cell Mol. Biol. 2017, 57, 15–17.
- Kuznetsov, A.V.; Margreiter, R.; Amberger, A.; Saks, V.; Grimm, M. Changes in mitochondrial redox state, membrane potential and calcium precede mitochondrial dysfunction in doxorubicin-induced cell death. Biochim. Biophys. Acta 2011, 1813, 1144–1152.
- Bonnet, S.; Michelakis, E.D.; Porter, C.; Andrade, M.; Thébaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; et al. An abnormal mitochondrial–hypoxia inducible factor-1α–Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats. Circulation 2006, 113, 2630–2641.
- Bonnet, S.; Rochefort, G.; Sutendra, G.; Archer, S.L.; Haromy, A.; Webster, L.; Hashimoto, K.; Michelakis, E.D. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc. Natl. Acad. Sci. USA 2007, 104, 11418–11423.
- McMurtry, M.S.; Archer, S.L.; Altieri, D.C.; Bonnet, S.; Haromy, A.; Harry, G.; Bonnet, S.; Puttagunta, L.; Michelakis, E.D. Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J. Clin. Investig. 2005, 115, 1479–1491.
- Hu, H.-L.; Zhang, Z.-X.; Chen, C.-S.; Cai, C.; Zhao, J.-P.; Wang, X. Effects of mitochondrial potassium channel and membrane potential on hypoxic human pulmonary artery smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 2010, 42, 661–666.
- Gaudry, M.J.; Jastroch, M. Molecular evolution of uncoupling proteins and implications for brain function. Neurosci. Lett. 2019, 696, 140–145.
- Boucherat, O.; Peterlini, T.; Bourgeois, A.; Nadeau, V.; Breuils-Bonnet, S.; Boilet-Molez, S.; Potus, F.; Meloche, J.; Chabot, S.; Lambert, C.; et al. Mitochondrial HSP90 accumulation promotes vascular remodeling in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 90–103.
- Sobolewski, A.; Rudarakanchana, N.; Upton, P.D.; Yang, J.; Crilley, T.K.; Trembath, R.; Morrell, N. Failure of bone morphogenetic protein receptor trafficking in pulmonary arterial hypertension: Potential for rescue. Hum. Mol. Genet. 2008, 17, 3180–3190.
- Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Sutendra, G.; Michelakis, E.D. Attenuating endoplasmic reticulum stress as a novel therapeutic strategy in pulmonary hypertension. Circulation 2013, 127, 115–125.
- Guignabert, C.; Bailly, S.; Humbert, M. Restoring BMPRII functions in pulmonary arterial hypertension: Opportunities, challenges and limitations. Expert Opin. Ther. Targets 2017, 21, 181–190.
- Zhuan, B.; Wang, X.; Wang, M.-D.; Li, Z.-C.; Yuan, Q.; Xie, J.; Yang, Z. Hypoxia induces pulmonary artery smooth muscle dysfunction through mitochondrial fragmentation-mediated endoplasmic reticulum stress. Aging 2020, 12, 23684–23697.
- Lindner, P.; Christensen, S.B.; Nissen, P.; Møller, J.V.; Engedal, N. Cell death induced by the ER stressor thapsigargin involves death receptor 5, a non-autophagic function of MAP1LC3B, and distinct contributions from unfolded protein response components. Cell Commun. Signal. 2020, 18, 12.
- Shore, G.C.; Papa, F.R.; Oakes, S.A. Signaling cell death from the endoplasmic reticulum stress response. Curr. Opin. Cell Biol. 2011, 23, 143–149.
- Yang, Y.-D.; Li, M.-M.; Xu, G.; Zhang, E.-L.; Chen, J.; Sun, B.; Chen, D.-W.; Gao, Y.-Q. Targeting mitochondria-associated membranes as a potential therapy against endothelial injury induced by hypoxia. J. Cell. Biochem. 2019, 120, 18967–18978.
- Siques, P.; Brito, J.; Pena, E. Reactive oxygen species and pulmonary vasculature during hypobaric hypoxia. Front. Physiol. 2018, 9, 865.
- Waypa, G.B.; Guzy, R.; Mungai, P.T.; Mack, M.M.; Marks, J.D.; Roe, M.W.; Schumacker, P.T. Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ. Res. 2006, 99, 970–978.
- Dunham-Snary, K.; Wu, D.; Potus, F.; Sykes, E.A.; Mewburn, J.D.; Charles, R.L.; Eaton, P.; Sultanian, R.A.; Archer, S.L. Ndufs2, a core subunit of mitochondrial complex I, is essential for acute oxygen-sensing and hypoxic pulmonary vasoconstriction. Circ. Res. 2019, 124, 1727–1746.
- Dunham-Snary, K.; Wu, D.; Sykes, E.A.; Thakrar, A.; Parlow, L.R.; Mewburn, J.D.; Parlow, J.L.; Archer, S.L. Hypoxic pulmonary vasoconstriction: From molecular mechanisms to medicine. Chest 2017, 151, 181–192.
- Li, X.-Q.; Zheng, Y.-M.; Reyes-García, J.; Wang, Y.-X. Diversity of ryanodine receptor 1-mediated Ca2+ signaling in systemic and pulmonary artery smooth muscle cells. Life Sci. 2021, 270, 119016.
- Liao, B.; Zheng, Y.-M.; Yadav, V.R.; Korde, A.S.; Wang, Y.-X. Hypoxia induces intracellular Ca2+ release by causing reactive oxygen species-mediated dissociation of FK506-binding protein 12.6 from ryanodine receptor 2 in pulmonary artery myocytes. Antioxid. Redox Signal. 2011, 14, 37–47.
- Wang, Y.-X.; Zheng, Y.-M. Role of ROS signaling in differential hypoxic Ca2+ and contractile responses in pulmonary and systemic vascular smooth muscle cells. Respir. Physiol. Neurobiol. 2010, 174, 192–200.
- Wang, Y.-X.; Zheng, Y.-M.; Mei, Q.-B.; Wang, Q.-S.; Collier, M.L.; Fleischer, S.; Xin, H.-B.; Kotlikoff, M.I. FKBP12.6 and cADPR regulation of Ca2+ release in smooth muscle cells. Am. J. Physiol. Cell Physiol. 2004, 286, C538–C546.
- Tang, W.-X.; Chen, Y.-F.; Zou, A.-P.; Campbell, W.B.; Li, P.-L. Role of FKBP12.6 in cADPR-induced activation of reconstituted ryanodine receptors from arterial smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1304–H1310.
- Korde, A.; Yadav, V.R.; Zheng, Y.-M.; Wang, Y.-X. Primary role of mitochondrial Rieske iron–sulfur protein in hypoxic ROS production in pulmonary artery myocytes. Free Radic. Biol. Med. 2011, 50, 945–952.
- Song, T.; Zheng, Y.-M.; Wang, Y.-X. Cross talk between mitochondrial reactive oxygen species and sarcoplasmic reticulum calcium in pulmonary arterial smooth muscle cells. Adv. Exp. Med. Biol. 2017, 967, 289–298.
- Wang, Y.-X.; Zheng, Y.-M. ROS-Dependent signaling mechanisms for hypoxic Ca2+ responses in pulmonary artery myocytes. Antioxid. Redox Signal. 2010, 12, 611–623.
- Grabacka, M.; Pierzchalska, M.; Dean, M.; Reiss, K. Regulation of ketone body metabolism and the role of PPARα. Int. J. Mol. Sci. 2016, 17, 2093.
- Nasser, S.; Vialichka, V.; Biesiekierska, M.; Balcerczyk, A.; Pirola, L. Effects of ketogenic diet and ketone bodies on the cardiovascular system: Concentration matters. World J. Diabetes 2020, 11, 584–595.
- Puchalska, P.; Crawford, P.A. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 2017, 25, 262–284.
- De Sa, H.A.; Chung, S.; Shaniuk, P.M. Sweet and salty: Diabetic ketoacidosis in a patient with nephrogenic diabetes insipidus. Cureus 2021, 13, e12682.
- Gibson, A.A.; Eroglu, E.I.; Rooney, K.; Harper, C.; McClintock, S.; Franklin, J.; Markovic, T.P.; Seimon, R.V.; Sainsbury, A. Urine dipsticks are not accurate for detecting mild ketosis during a severely energy restricted diet. Obes. Sci. Pract. 2020, 6, 544–551.
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52.
- Arima, Y.; Nakagawa, Y.; Takeo, T.; Ishida, T.; Yamada, T.; Hino, S.; Nakao, M.; Hanada, S.; Umemoto, T.; Suda, T.; et al. Murine neonatal ketogenesis preserves mitochondrial energetics by preventing protein hyperacetylation. Nat. Metab. 2021, 3, 196–210.
- Dhillon, K.K.; Gupta, S. Biochemistry, ketogenesis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–426.
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose tissue remodeling: Its role in energy metabolism and metabolic disorders. Front. Endocrinol. 2016, 7, 30.
- Kadochi, Y.; Mori, S.; Fujiwara-Tani, R.; Luo, Y.; Nishiguchi, Y.; Kishi, S.; Fujii, K.; Ohmori, H.; Kuniyasu, H. Remodeling of energy metabolism by a ketone body and medium-chain fatty acid suppressed the proliferation of CT26 mouse colon cancer cells. Oncol. Lett. 2017, 14, 673–680.
- Serviddio, G.; Giudetti, A.M.; Bellanti, F.; Priore, P.; Rollo, T.; Tamborra, R.; Siculella, L.; Vendemiale, G.; Altomare, E.; Gnoni, G.V. Oxidation of hepatic carnitine palmitoyl transferase-I (CPT-I) impairs fatty acid beta-oxidation in rats fed a methionine-choline deficient diet. PLoS ONE 2011, 6, e24084.
- Latruffe, N. Human peroxisomal 3-ketoacyl-CoA thiolase: Tissue expression and metabolic regulation: Human peroxisomal thiolase. Adv. Exp. Med. Biol. 2020, 1299, 161–167.
- Galluzzi, L.; Kroemer, G. Aberrant ketolysis fuels hepatocellular cancer progression. Cell Res. 2016, 26, 1077–1078.
- Dumbrepatil, A.B.; Zegalia, K.A.; Sajja, K.; Kennedy, R.T.; Marsh, E.N.G. Targeting viperin to the mitochondrion inhibits the thiolase activity of the trifunctional enzyme complex. J. Biol. Chem. 2020, 295, 2839–2849.
- Fletcher, J.A.; Deja, S.; Satapati, S.; Fu, X.; Burgess, S.C.; Browning, J.D. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 2019, 5, e127737.
- Likhodii, S.S.; Musa, K.; Cunnane, S.C. Breath acetone as a measure of systemic ketosis assessed in a rat model of the ketogenic diet. Clin. Chem. 2002, 48, 115–120.
- Musa-Veloso, K.; Likhodii, S.S.; Cunnane, S.C. Breath acetone is a reliable indicator of ketosis in adults consuming ketogenic meals. Am. J. Clin. Nutr. 2002, 76, 65–70.
- Chen, F.; Qian, L.-H.; Deng, B.; Liu, Z.-M.; Zhao, Y.; Le, Y.-Y. Resveratrol protects vascular endothelial cells from high glucose-induced apoptosis through inhibition of NADPH oxidase activation-driven oxidative stress. CNS Neurosci. Ther. 2013, 19, 675–681.
- Zhu, M.; Chen, J.; Jiang, H.; Miao, C. Propofol protects against high glucose-induced endothelial adhesion molecules expression in human umbilical vein endothelial cells. Cardiovasc. Diabetol. 2013, 12, 13.
- Grinnan, D.; Farr, G.; Fox, A.; Sweeney, L. The role of hyperglycemia and insulin resistance in the development and progression of pulmonary arterial hypertension. J. Diabetes Res. 2016, 2016, 2481659.
- La Frano, M.R.; Fahrmann, J.F.; Grapov, D.; Pedersen, T.L.; Newman, J.; Fiehn, O.; Underwood, M.A.; Mestan, K.K.; Steinhorn, R.H.; Wedgwood, S. Umbilical cord blood metabolomics reveal distinct signatures of dyslipidemia prior to bronchopulmonary dysplasia and pulmonary hypertension. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2018, 315, L870–L881.
- Kim, D.; Roberts, C.; McKenzie, A.; George, M.P. Nutritional ketosis to treat pulmonary hypertension associated with obesity and metabolic syndrome: A case report. Pulm. Circ. 2021, 11, 2045894021991426.
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724.
- Kim, H.-I.; Ahn, Y.-H. Role of peroxisome proliferator-activated receptor-γ in the glucose-sensing apparatus of liver and β-cells. Diabetes 2004, 53, S60–S65.
- Semple, R.K.; Chatterjee, V.K.K.; O’Rahilly, S. PPAR gamma and human metabolic disease. J. Clin. Investig. 2006, 116, 581–589.
- Wang, Y.-X. PPARs: Diverse regulators in energy metabolism and metabolic diseases. Cell Res. 2010, 20, 124–137.
- Yang, Q.; Long, Q. PPARδ, a potential therapeutic target for heart disease. Nucl. Recept. Res. 2018, 5, 101375.
- Hansmann, G.; Calvier, L.; Risbano, M.G.; Chan, S.Y. Activation of the metabolic master regulator PPARγ: A potential pioneering therapy for pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2020, 62, 143–156.
- Green, D.E.; Murphy, T.C.; Kang, B.-Y.; Searles, C.D.; Hart, C.M. PPARγ ligands attenuate hypoxia-induced proliferation in human pulmonary artery smooth muscle cells through modulation of microRNA-21. PLoS ONE 2015, 10, e0133391.
- Reddy, A.; Lakshmi, S.; Kleinhenz, J.M.; Sutliff, R.L.; Hart, C.M.; Reddy, R.C. Endothelial cell peroxisome proliferator–activated receptor γ reduces endotoxemic pulmonary inflammation and injury. J. Immunol. 2012, 189, 5411–5420.