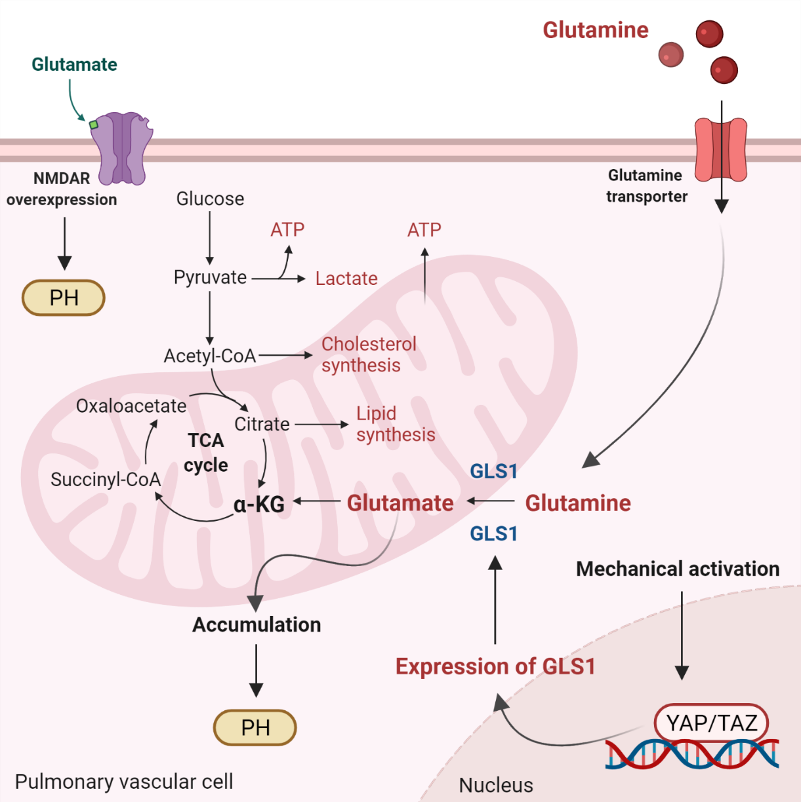
Ketone bodies, or simply ketones, are highly polar molecules produced by β-oxidation of fatty acids in the mitochondria of the liver cells. However, these molecules may be produced by enterocytes, astrocytes, and kidney ECs to a lesser extent [
24,
26,
143]. Ketones are produced in response to reduced glucose availability, e.g., during periods of prolonged fasting, high-performance exercise, or a pathophysiological state, such as type I diabetes [
144,
145]. It has been postulated that patients with PAH have reduced oral glucose tolerance and lipid and ketone metabolism predominate over the glucose control [
58].
Ketone’s metabolism is divided in ketogenesis and ketolysis. Ketogenesis, which mainly occurs in perivenous hepatocytes, produces three molecules: acetone, acetoacetate, and β-HB [
146], which represents the most abundant ketone body [
147,
148,
149]. It is well known that adipocytes store great amounts of energy as fatty acids [
150]. When fasting or exercising, glycogen stores are used in the beginning. Once glycogen is depleted, fatty acids from adipocytes are transferred into the liver by the enzyme carnitine palmitoyltransferase (CPT-1), where they are metabolized in the mitochondria to form ketone bodies [
151,
152]. These lipid derivatives enter the systemic circulation and reach highly metabolic tissues, e.g., muscles and nervous system, which convert ketones into acetyl coenzyme A (acetyl-CoA) for alternative energy metabolism. The detailed process occurs as follows: two molecules of acetyl-CoA are biotransformed in acetoacetyl-CoA by the action of the acetyl-CoA acetyltransferase (ACAT), a thiolase [
153]. Then, 3-hydroxy-3-methylglutaryl Co-A synthase (HMGS2) condenses acetyl-CoA with acetoacetyl-CoA to produce HMG-CoA. Afterward, HMG-CoA is broken into acetoacetate via HMG-CoA lyase (HMGCL). Finally, acetoacetate is further bioconverted to acetone (by decarboxylation) or to β-HB by the action of 3-hydroxybutyrate dehydrogenase (BDH1) [
148].
On the other hand, throughout ketolysis, acetoacetate and β-HB are used as an energy source by the mitochondria of several extrahepatic tissues [
148,
154]. β-HB is transformed to acetoacetate by the BDH1, and this last product is turned into acetoacetyl-CoA by the enzyme beta-ketoacyl-CoA transferase (OXCT1 or SCOT). Acetoacetyl-CoA is separated by thiolase in two molecules of acetyl-CoA [
155], that enters the TCA, and subsequently the oxidative phosphorylation, resulting in the generation of ATP [
148,
156]. Importantly, acetone (the other ketone body) cannot be biotransformed into acetyl-CoA and ends up eliminated through urine or exhaled [
157,
158].
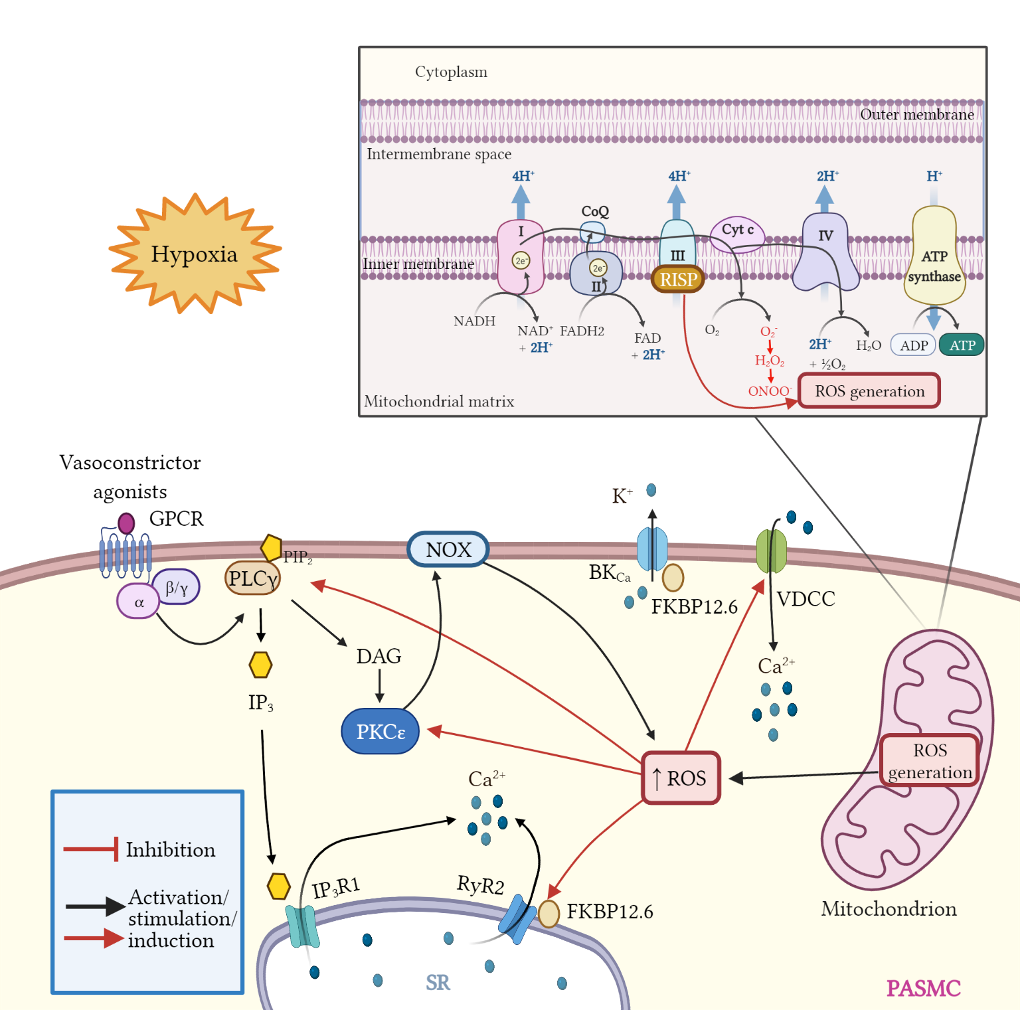
High glucose levels may elicit the over production of ROS, mainly through NOX4 activity in endothelial cells [
161]. In addition, in these cells, hyperglycemia triggers NF-κB signaling pathway leading to the upregulation of proinflammatory cytokines and endothelial adhesion molecules [
162]. The induction of endothelial selectin (E-selectin), intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and increased mononuclear-endothelial adhesion in addition to ROS generation and NF-κB activity increase vascular permeability and facilitates endothelial barrier dysfunction [
162].
The role of ketones in the development of PH remains elusive. In this regard, the evidence indicates a relationship between the RVF occurring in PH and metabolic disorders (risk factors/comorbidities in human PH), such as high blood sugar [
58], insulin resistance [
164], dyslipidemia [
165], and abdominal obesity [
166].
It is well known that 70–90% of ATP produced in heart comes from the oxidation of FA in mitochondria. The residual percentage is generated by the metabolism of glucose, ketone bodies and amino acids [
171]. In this context, Peroxisome proliferator-activated receptor γ (PPARγ) has been shown to regulate glucose and FA metabolism in adipocytes, hepatocytes, skeletal muscle, and pancreatic β cells [
172,
173,
174]. PPARs belong to the superfamily of nuclear receptors serving as ligand-activated transcription factors. This receptor subfamily is composed of three members, PPARα, PPARγ and PPARδ, which combine with retinoid X receptors (RXRs) forming heterodimers and binding to specific DNA sites to promote genic transcription [
173]. PPARγ is a master regulator of adipogenesis expressed mostly in adipose tissue and liver, as well as PPARα. PPARδ is ubiquitously expressed and all three subtypes are expressed in heart [
175]. Emerging evidence highlights that PPARγ acts as a strong, protective regulator in PAH [
176], PASMCs [
177], and PAECs [
178]. As it concerns, Legchenko et al., in a SU5416/hypoxia-induced PAH rat model, demonstrated that the oral administration of pioglitazone (a PPARγ agonist) fully abolishes severe PAH and vascular remodeling, and prevents RVF (Figure
3).
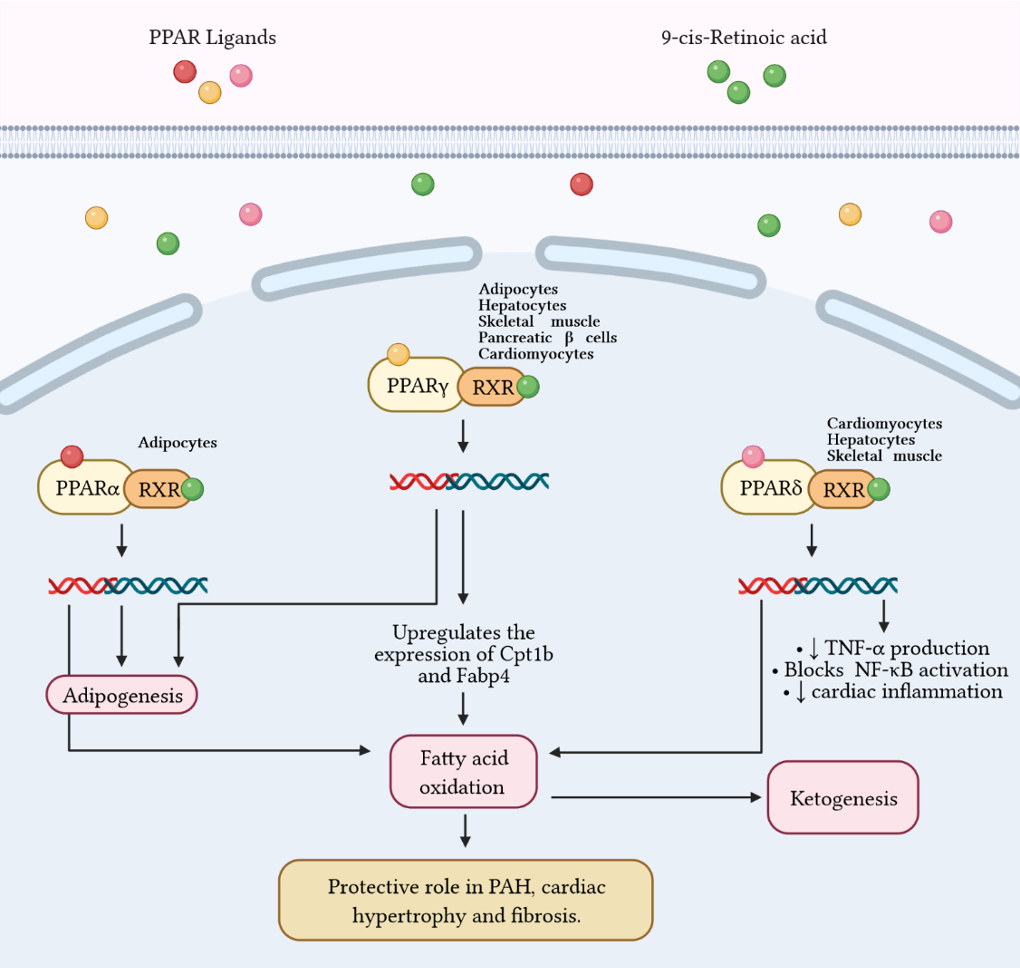
Figure 3 Fatty acid metabolism and its byproducts (ketones) play an important role in the development of right ventricular failure (RVF) and pulmonary arterial hypertension (PAH). Peroxisome proliferator-activated receptors (PPAR) belong to the superfamily of nuclear receptors that serve as ligand-activated transcription factors and consist of three members, PPARa, PPARg, and PPARd. These receptors together with retinoid X receptors (RXR) form heterodimers and bind to specific DNA sites to promote genetic transcription. PPARa is a master regulator of adipogenesis expressed mainly in adipose tissue and liver, as is PPARg. Additionally, PPARg regulates glucose and fatty acid metabolism in adipocytes, hepatocytes, skeletal muscle, and pancreatic b-cells. PPARg agonists, such as pioglitazone, increase the expression of Cpt1b and Fabp4, proteins involved in fatty acid oxidation and transport in cardiomyocytes. These effects favor mitochondrial fatty acid oxidation and ATP production, leading to reversal of cardiac hypertrophy, fibrosis, and eliminating severe PAH. Furthermore, PPARd stimulates fatty acid oxidation, decreases right ventricle hypertrophy and pulmonary congestion. In cardiac inflammation, PPARd blocks nuclear factor kB (NF-kB) activation and inhibits tumor necrosis factor (TNF)-a synthesis.
8. Conclusions
Mitochondria are involved in essential cellular regulatory and homeostatic process in the cardiovascular system and particularly in VSMCs. Mitochondrial dysfunction has been widely associated with several diseases including PH. These organelles exert a strict control of ROS and ketones production. Uncontrolled ROS generation and its crosstalk with Ca2+ signaling have been shown to contribute and aggravate pulmonary hypertension. Thus, anti-ROS therapies targeting implicated proteins such as RISP should be investigated as novel alternatives for the treatment of this ailment. On the other hand, ketone bodies seem to offer a protection against oxidative stress damage in vitro and in vivo; however, they also may exert anti-inflammatory or pro-inflammatory roles in the cardiovascular system, and further research is needed to understand their different roles. Collectively, a better comprehension of the unique roles of RISP-dependent mitochondrial ROS and their specific interactions with mitochondrial Ca2+ signaling, NF-κB-mediated inflammatory responses, and ketone-associated oxidative stress can significantly improve our understanding of the molecular pathogenesis of PH and associated RVF. This new knowledge may also help to develop and implement innovative therapies in the treatment of PH and other vascular diseases.