 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Lurdes Santos Cristiano | -- | 2655 | 2022-06-30 08:53:28 | | | |
| 2 | Peter Tang | Meta information modification | 2655 | 2022-06-30 09:27:21 | | | | |
| 3 | Peter Tang | -3 word(s) | 2652 | 2022-07-04 08:13:59 | | |
Video Upload Options
Pyrazoles are known as versatile scaffolds in organic synthesis and medicinal chemistry, often used as starting materials for the preparation of more complex heterocyclic systems with relevance in the pharmaceutical field. Pyrazoles are also interesting compounds from a structural viewpoint, mainly because they exhibit tautomerism. This phenomenon may influence their reactivity, with possible impact on the synthetic strategies where pyrazoles take part, as well as on the biological activities of targets bearing a pyrazole moiety, since a change in structure translates into changes in properties. Investigations of the structure of pyrazoles that unravel the tautomeric and conformational preferences are therefore of upmost relevance. 3(5)-Aminopyrazoles are largely explored as precursors in the synthesis of condensed heterocyclic systems, namely pyrazolo[1,5-a]pyrimidines.
1. Introduction
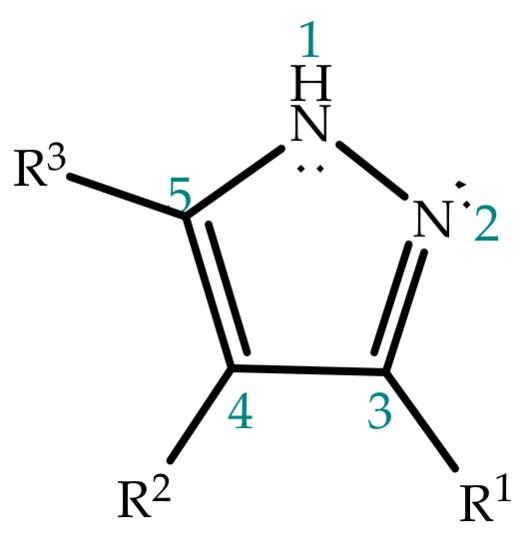
2. Pyrazole Properties
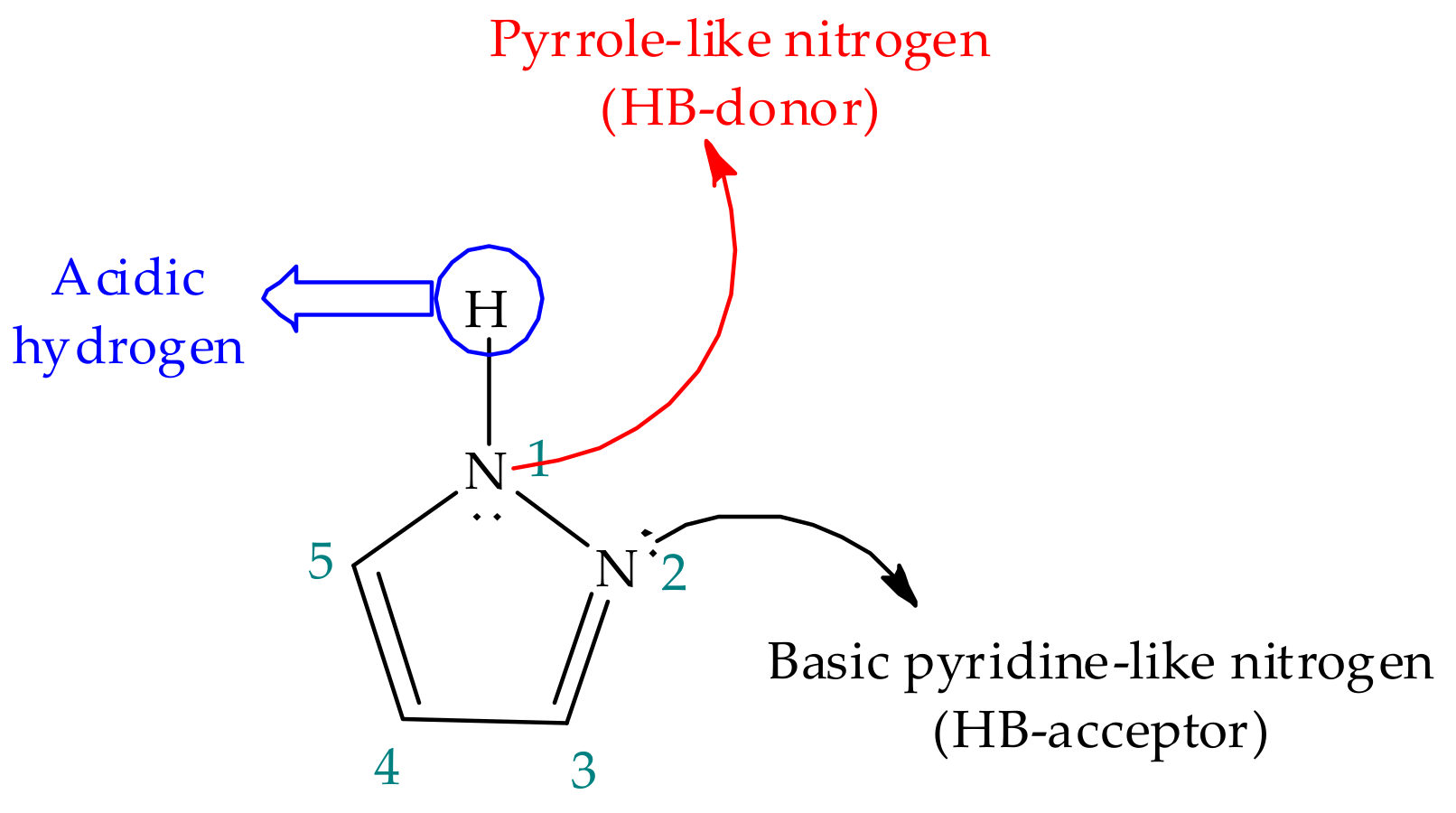
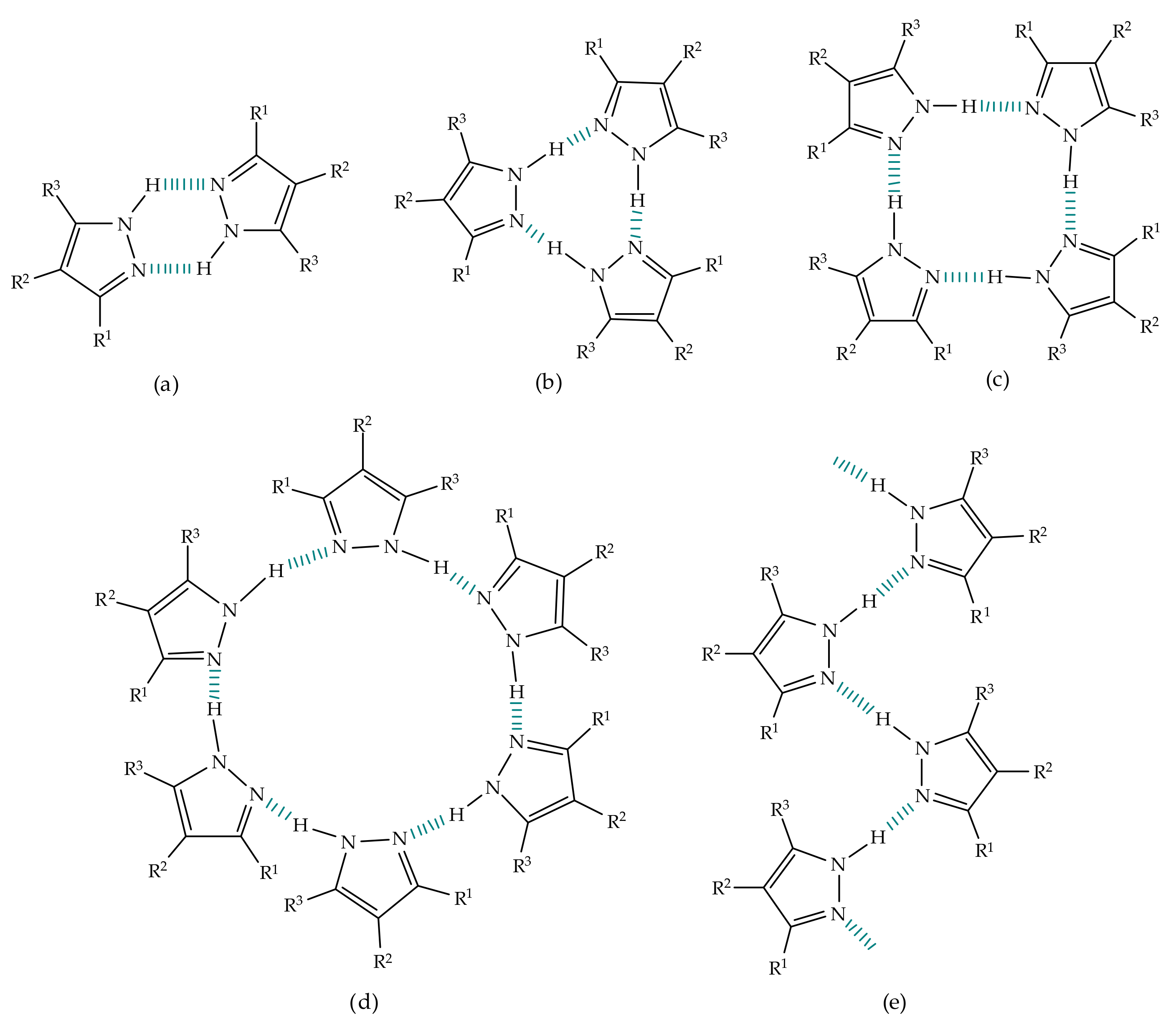
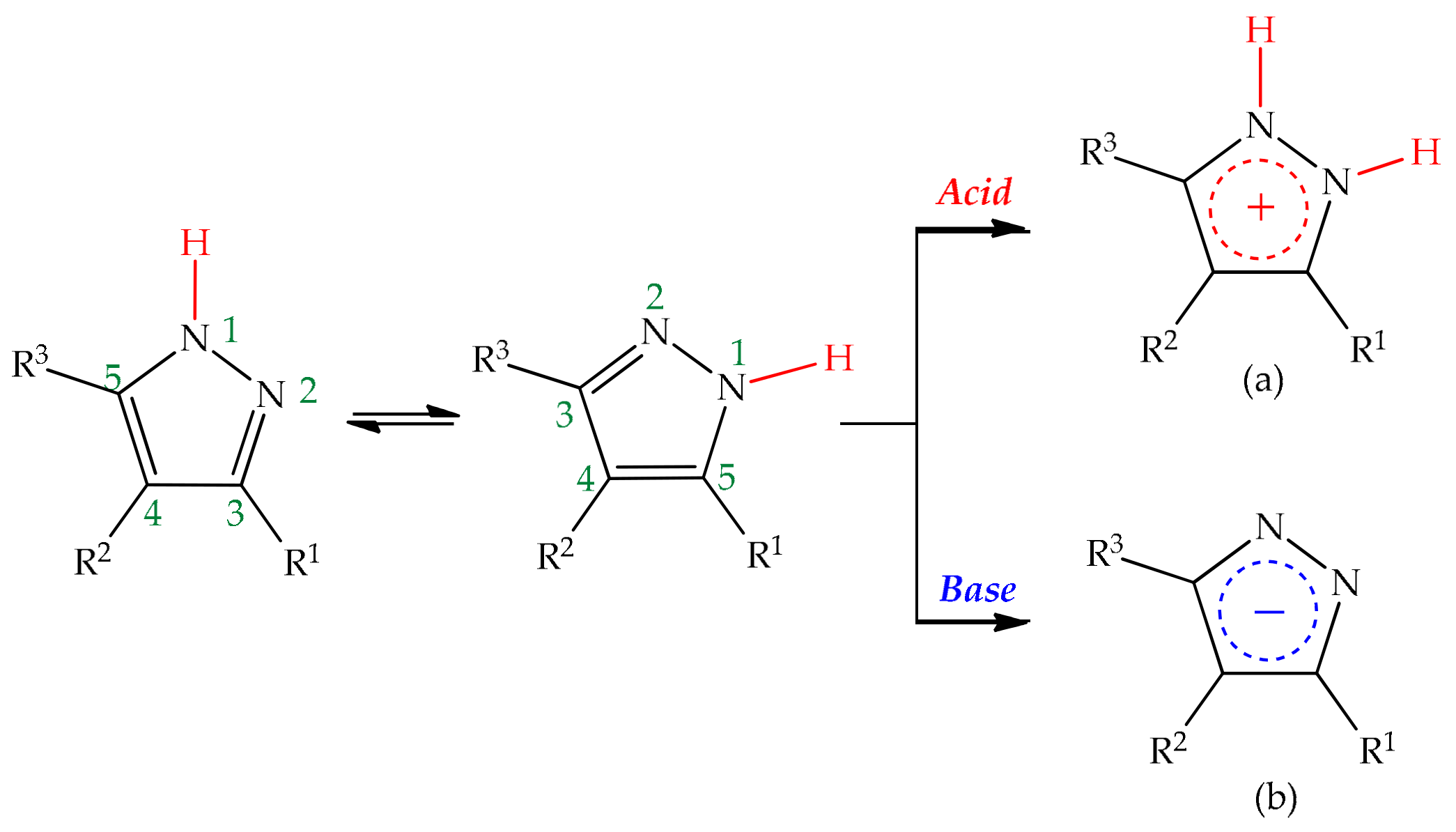
3. Tautomerism in Pyrazoles
4. Pyrazoles in Organic Synthesis
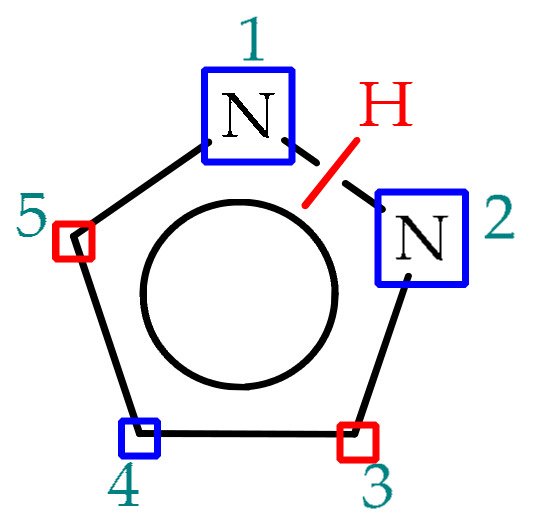
5. 3(5)-Aminopyrazole
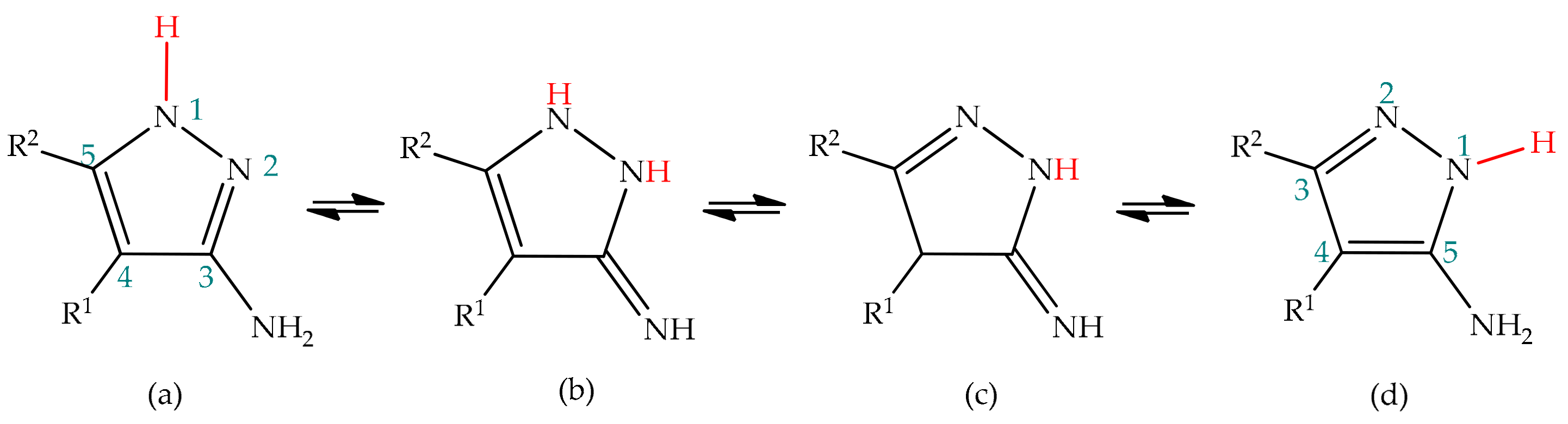
5.1. Tautomerism in 3(5)-Aminopyrazole
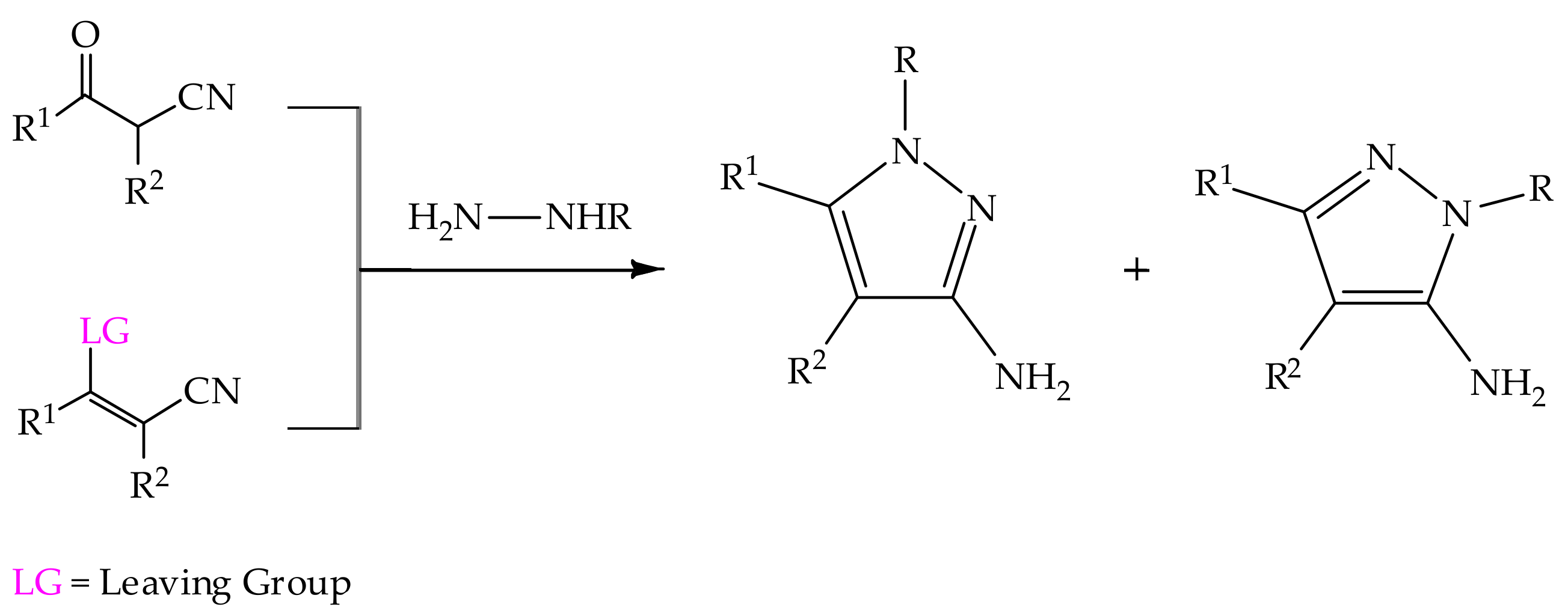
5.2. 3(5)-Aminopyrazole in Heterocyclic Synthesis
References
- Schofield, K.; Grimmett, M.R.; Grimmett, M.R.; Keene, B.R. Heteroaromatic Nitrogen Compounds: The Azoles; Cambridge University Press: Cambridge, UK, 1976.
- Andes, D.R.; Dismukes, W.E. Azoles. In Essentials of Clinical Mycology, 2nd ed.; Kauffman, C.A., Pappas, P.G., Sobel, J.D., Dismukes, W.E., Eds.; Springer: New York, NY, USA, 2011; pp. 61–93.
- Kumar, S.S.; Kavitha, H.P. Synthesis and Biological Applications of Triazole Derivatives—A Review. Mini. Rev. Org. Chem. 2013, 10, 40–65.
- Wei, C.X.; Bian, M.; Gong, G.H. Tetrazolium compounds: Synthesis and applications in medicine. Molecules 2015, 20, 5528–5553.
- Bai, S.Q.; Young, D.J.; Hor, T.S.A. Nitrogen-rich azoles as ligand spacers in coordination polymers. Chem. Asian J. 2011, 6, 292–304.
- Katritzky, A.R.; Ramsden, C.A.; Joule, J.A.; Zhdankin, V.V. Reactivity of Five-membered Rings with Two or More Heteroatoms. In Handbook of Heterocyclic Chemistry, 3rd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2010; pp. 473–604.
- Behr, L.C.; Fusco, R.; Jarboe, C.H. Pyrazoles, Pyrazolines, Pyrazolidines, Indazoles and Condensed Rings; John Wiley & Sons: New York, NY, USA, 1967; p. 150.
- Kumar, V.; Kaur, K.; Gupta, G.K.; Sharma, A.K. Pyrazole containing natural products: Synthetic preview and biological significance. Eur. J. Med. Chem. 2013, 69, 735–753.
- Vicentini, C.B.; Mares, D.; Tartari, A.; Manfrini, M.; Forlani, G. Synthesis of Pyrazole Derivatives and Their Evaluation as Photosynthetic Electron Transport Inhibitors. J. Agric. Food Chem. 2004, 52, 1898–1906.
- El-Mekabaty, A. Utility of 5-amino-1-phenyl-1H-pyrazole-4-carboxamide in heterocyclic synthesis. Synth. Commun. 2014, 44, 875–896.
- Kumari, S.; Paliwal, S.; Chauhan, R. Synthesis of pyrazole derivatives possessing anticancer activity: Current status. Synth. Commun. 2014, 44, 1521–1578.
- Katritzky, A.R. Advances in Heterocyclic Chemistry 76; Academic Press: San Diego, CA, USA, 2000.
- Shaabani, A.; Nazeri, M.T.; Afshari, R. 5-Amino-pyrazoles: Potent reagents in organic and medicinal synthesis. Mol. Divers. 2019, 23, 751–807.
- Elmaati, T.M.A.; El-Taweel, F.M. New Trends In The Chemistry Of 5-Aminopyrazoles. J. Heterocycl. Chem. 2004, 41, 109–134.
- Elnagdi, M.H.; Elmoghayar, M.R.H.; Elgemeie, G.E.H. Chemistry of Pyrazolopyrimidines. Adv. Heterocycl. Chem. 1987, 41, 319–376.
- Salem, M.A.; Helal, M.H.; Gouda, M.A.; EL-Gawad, H.H.A.; Shehab, M.A.M.; El-Khalafawy, A. Recent synthetic methodologies for pyrazolopyrimidine. Synth. Commun. 2019, 49, 1750–1776.
- Cherukupalli, S.; Karpoormath, R.; Chandrasekaran, B.; Hampannavar, G.A.; Thapliyal, N.; Palakollu, V.N. An insight on synthetic and medicinal aspects of pyrazolo pyrimidine scaffold. Eur. J. Med. Chem. 2017, 126, 298–352.
- Mohamed, M.S.; Mahmoud, A.M. Believes versus evidence-based regio-orientation in the structure assignment of pyrazolo pyrimidines. J. Appl. Pharm. Sci. 2019, 9, 126–144.
- Brown, A.W. Recent Developments in the Chemistry of Pyrazoles, 1st ed.; Elsevier Inc.: San Diego, CA, USA, 2018; Volume 126.
- Anwar, H.F.; Elnagdi, M.H. Recent developments in aminopyrazole chemistry. ARKIVOC 2009, 2009, 198–250.
- Mert, S.; Kasimogullari, R.; Ok, S. A Short Review on Pyrazole Derivatives and their Applications. J. Postdr. Res. 2014, 2, 64–72.
- Kalaria, P.N.; Karad, S.C.; Raval, D.K. A review on diverse heterocyclic compounds as the privileged scaffolds in antimalarial drug discovery. Eur. J. Med. Chem. 2018, 158, 917–936.
- Kumar, H.; Saini, D.; Jain, S.; Jain, N. Pyrazole scaffold: A remarkable tool in the development of anticancer agents. Eur. J. Med. Chem. 2013, 70, 248–258.
- Faria, J.V.; Vegi, P.F.; Miguita, A.G.C.; Santos, M.S.d.; Boechat, N.; Bernardino, A.M.R. Recently reported biological activities of pyrazole compounds. Bioorg. Med. Chem. 2017, 25, 5891–5903.
- Makino, K.; Kim, H.S.; Kurasawa, Y. Synthesis of pyrazoles and condensed pyrazoles. J. Heterocycl. Chem. 1999, 36, 321–332.
- Schmidt, A.; Dreger, A. Recent Advances in the Chemistry of Pyrazoles. Properties, Biological Activities, and Syntheses. Curr. Org. Chem. 2011, 15, 1423–1463.
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-Aizari, F.A.; Ansar, M. Synthesis and pharmacological activities of Pyrazole derivatives: A review. Molecules 2018, 23, 134.
- Alam, M.J.; Alam, O.; Alam, P.; Naim, M.J. A Review on Pyrazole chemical entity and Biological Activity. Int. J. Pharma Sci. Res. 2015, 6, 1433–1442.
- Kumar, K.A.; Jayaroopa, P. Pyrazoles: Synthetic strategies and their pharmaceutical applications-an overview. Int. J. PharmTech Res. 2013, 5, 1473–1486.
- Chauhan, A.; Sharma, P.K.; Kaushik, N. Pyrazole: A versatile moiety. Int. J. ChemTech Res. 2011, 3, 11–17.
- Bekhit, A.A.; Hymete, A.; Bekhit, A.E.A.; Damtew, A.; Aboul-Enein, H.Y. Pyrazoles as Promising Scaffold for the Synthesis of Anti-Inflammatory and/or Antimicrobial Agent: A Review. Mini Rev. Med. Chem. 2012, 10, 1014–1033.
- Nimbalkar, S.; Hote, S.V. Pyrazole Derivatives and their Synthesis—A review. Int. J. Recent Innov. Trends Comput. Commun. 2015, 3, 61–65.
- Fustero, S.; María, S.; Barrio, P.; Sim, A. From 2000 to Mid-2010 : A Fruitful Decade for the Synthesis of Pyrazoles. Chem. Rev. 2011, 111, 6984–7034.
- Giller, S.A.; Mazheika, I.B.; Grandberg, I.I. Electron density distribution in heterocyclic systems with two vicinal nitrogen atoms. II. Pyrazole dipole moments. Khimiya Geterotsiklicheskikh Soedin. 1965, 1, 103–106.
- Chermahini, A.N.; Teimouri, A.; Beni, A.S.; Dordahan, F. Theoretical studies on the effect of substituent in the proton transfer reaction of 4-substituted pyrazoles. Comput. Theor. Chem. 2013, 1008, 67–73.
- Catalán, J.; Sánchez-Cabezudo, M.; de Paz, J.L.G.; Elguero, J.; Taft, R.W.; Anvia, F. The tautomerism of 1,2,3-triazole, 3(5)-methylpyrazole and their cations. J. Comput. Chem. 1989, 10, 426–433.
- Catalan, J.; Abboud, J.L.M.; Elguero, J. Basicity and Acidity of Azoles. Adv. Heterocycl. Chem. 1987, 41, 187–274.
- Castaneda, J.P.; Denisov, G.S.; Kucherov, S.Y.; Schreiber, V.M.; Shurukhina, A.V. Infrared and ab initio studies of hydrogen bonding and proton transfer in the complexes formed by pyrazoles. J. Mol. Struct. 2003, 660, 25–40.
- Claramunt, R.M.; López, C.; María, M.D.S.; Sanz, D.; Elguero, J. The use of NMR spectroscopy to study tautomerism. Prog. Nucl. Magn. Reson. Spectrosc. 2006, 49, 169–206.
- Pérez, J.; Riera, L. Pyrazole complexes and supramolecular chemistry. Eur. J. Inorg. Chem. 2009, 33, 4913–4925.
- Claramunt, R.M.; Cornago, P.; Torres, V.; Pinilla, E.; Torres, M.R.; Samat, A.; Lokshin, V.; Valés, M.; Elguero, J. The structure of pyrazoles in the solid state: A combined CPMAS, NMR, and crystallographic study. J. Org. Chem. 2006, 71, 6881–6891.
- Alkorta, I.; Goya, P.; Elguero, J.; Singh, S.P. A simple approach to the tautomerism of aromatic heterocycles. Natl. Acad. Sci. Lett. 2007, 30, 139–159.
- Chermahini, A.N.; Teimouri, A. Theoretical studies on proton transfer reaction of 3(5)-substituted pyrazoles. J. Chem. Sci. 2014, 126, 273–281.
- Oziminski, W.P. The kinetics of water-assisted tautomeric 1,2-proton transfer in azoles: A computational approach. Struct. Chem. 2016, 27, 1845–1854.
- Knorr, L. Ueber Abkömmlinge der Phenolform des 1-Phenyl-3-methyl-5-pyrazolons. Eur. J. Inorg. Chem. 1895, 28, 706–714.
- Elattar, K.M.; Fadda, A.A. Chemistry of antipyrine. Synth. Commun. 2016, 46, 1567–1594.
- Katritzky, A.R.; Hall, C.D.; El-Gendy, B.E.D.M.; Draghici, B. Tautomerism in drug discovery. J. Comput. Aided. Mol. Des. 2010, 24, 475–484.
- Taylor, P.J.; van der Zwan, G.; Antonov, L. Tautomerism: Introduction, History, and Recent Developments in Experimental and Theoretical Methods. In Tautomerism: Methods and Theories, 1st ed.; Antonov, L., Ed.; Wiley-VCH: Weinheim, Germany, 2014; pp. 1–24.
- Raczyńska, E.D.; Kosińska, W.; Ośmiałowski, B.; Gawinecki, R. Tautomeric equilibria in relation to Pi-electron delocalization. Chem. Rev. 2005, 105, 3561–3612.
- Minkin, V.I.; Garnovskii, A.D.; Elguero, J.; Katritzky, A.R.; Denisko, O.V. Tautomerism of Heterocycles: Five-Membered Rings with Two or More Heteroatoms. In Advances in Heterocyclic Chemistry; Katritzky, A.R., Ed.; Academic Press: San Diego, CA, USA, 2000; pp. 159–323.
- Minkin, V.I.; Olekhnovich, L.P.; Zhdanov, Y.A. Molecular Design of Tautomeric Compounds; D. Reidel Publishing Company: Dordrecht, The Netherlands, 1981; Volume 14.
- Elguero, J.; Katritzky, A.R.; Denisko, O.V. Prototropic Tautomerism of Heterocycles: Heteroaromatic Tautomerism—General Overview and Methodology. In Advances in Heterocyclic Chemistry; Katritzky, A.R., Ed.; Academic Press: San Diego, CA, USA, 2000; pp. 2–84.
- Martin, Y.C. Let’s not forget tautomers. J. Comput. Aided. Mol. Des. 2009, 23, 693–704.
- Alkorta, I.; Elguero, J. 1,2-Proton shifts in pyrazole and related systems: A computational study of -sigmatropic migrations of hydrogen and related phenomena. J. Chem. Soc. Perkin Trans. 1998, 2, 2497–2504.
- Hall, D.G. Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine; WILEY-VCH: Weinheim, Germany, 2005.
- McLaughlin, M.; Marcantonio, K.; Chen, G.Y.; Davies, I.W. A simple, modular method for the synthesis of 3,4,5-trisubstituted pyrazoles. J. Org. Chem. 2008, 73, 4309–4312.
- Huang, A.; Wo, K.; Lee, S.Y.C.; Kneitschel, N.; Chang, J.; Zhu, K.; Mello, T.; Bancroft, L.; Norman, N.J.; Zheng, S.L. Regioselective Synthesis, NMR, and Crystallographic Analysis of N1-Substituted Pyrazoles. J. Org. Chem. 2017, 82, 8864–8872.
- Kost, A.N.; Grandberg, I.I. Progress in Pyrazole Chemistry. Adv. Heterocycl. Chem. 1966, 6, 347–429.
- Halcrow, M.A. Pyrazoles and pyrazolides—Flexible synthons in self-assembly. J. Chem. Soc. Dalt. Trans. 2009, 9226, 2059–2073.
- Aggarwal, R.; Kumar, S. 5-Aminopyrazole as precursor in design and synthesis of fused pyrazoloazines. Beilstein J. Org. Chem. 2018, 14, 203–242.
- Kumar, A.; Paliwal, D.; Saini, D.; Thakur, A.; Aggarwal, S.; Kaushik, D. A comprehensive review on synthetic approach for antimalarial agents. Eur. J. Med. Chem. 2014, 85, 147–178.
- Puello, J.Q. Structure and tautomerism of 3(5)-amino-5(3)-arylpyrazoles in the solid state and in solution: An X-ray and NMR study. Tetrahedron 1997, 53, 10783–10802.
- Kusakiewicz-Dawid, A.; Porada, M.; Dziuk, B.; Siodlak, D. Annular Tautomerism of 3(5)-Disubstituted-1H-pyrazoles with Ester and Amide Groups. Molecules 2019, 24, 2632.
- Begtrup, M.; Boyer, G.; Cabildo, P.; Cativiela, C.; Claramunt, R.M.; Elguero, J.; García, J.I.; Toiron, C.; Vedsø, P. 13C NMR of pyrazoles. Magn. Reson. Chem. 1993, 31, 107–168.
- Gonzalez, E.; Faure, R.; Vincent, E.-J.; Espada, M.; Elguero, J. Etude en Résonance Magnétique Nucléaire du Carbone-13 de Quelques Aminopyrazoles. Sur le Problème de la Détermination des Constantes d’Equilibre Tautomère. Org. Magn. Reson. 1979, 12, 587–592.
- Dorn, H. Tautomerie und Nomenklatur der ‘Pyrazolone’ und Amino-pyrazole. J. Für Prakt. Chem. 1973, 315, 382–418.
- Lim, F.P.L.; Tan, K.C.; Luna, G.; Tiekink, E.R.T.; Dolzhenko, A.V. A new practical synthesis of 3-amino-substituted 5-aminopyrazoles and their tautomerism. Tetrahedron 2019, 75, 2314–2321.
- Larina, L.I. Tautomerism and Structure of Azoles: Nuclear Magnetic Resonance Spectroscopy. In Advances in Heterocyclic Chemistry; Scriven, E.F.V., Ramsden, C.A., Eds.; Elsevier Ltd.: San Diego, CA, USA, 2018; Volume 126, pp. 233–321.
- Catalán, J.; Menéndez, M.; Laynez, J.; Claramunt, R.M.; Bruix, M.; De Mendoza, J.; Elguero, J. Basicity of azoles. Basicity of C-Aminopyrazoles in Relation to Tautomeric and Protonation Studies. J. Heterocycl. Chem. 1985, 22, 997–1000.
- Marín-Luna, M.; Alkorta, I.; Elguero, J. A theoretical study of the gas phase (proton affinity) and aqueous (pKa) basicity of a series of 150 pyrazoles. New J. Chem. 2015, 39, 2861–2871.
- Fichez, J.; Busca, P.; Prestat, G. Recent advances in aminopyrazoles synthesis and functionalization. Targets Heterocycl. Syst. 2017, 21, 322–347.
- El-Sattar, N.E.A.A.; Badawy, E.H.K.; Abdel-Mottaleb, M.S.A. Synthesis of Some Pyrimidine, Pyrazole, and Pyridine Derivatives and Their Reactivity Descriptors. J. Chem. 2018, 1–11.
- Trujillo, C.; Sánchez-Sanz, G.; Alkorta, I.; Elguero, J. Computational Study of Proton Transfer in Tautomers of 3- and 5-Hydroxypyrazole Assisted by Water. ChemPhysChem 2015, 16, 2140–2150.
- Kruszyk, M.; Jessing, M.; Kristensen, J.L.; Jørgensen, M. Computational Methods to Predict the Regioselectivity of Electrophilic Aromatic Substitution Reactions of Heteroaromatic Systems. J. Org. Chem. 2016, 81, 5128–5134.
- Jarończyk, M.; Dobrowolski, J.C.; Mazurek, A.P. Theoretical studies on tautomerism and IR spectra of pyrazole derivatives. J. Mol. Struct. THEOCHEM 2004, 673, 17–28.


