The observation that pyrazoles are very rare in nature was ascribed to the presence of a single (-N-N-) bond in their structure, a chemical motif believed to be of very difficult formation by living organisms [
8]. Hence, pyrazoles are generally of synthetic origin and serve as building blocks in the synthesis of many other heterocyclic systems, most of which are biologically active, and interesting from a medicinal point of view, rendering this class of compounds worthy of deeper investigations [
9,
10,
11].
The rich reactivity of pyrazoles is linked to their challenging structure, with the possibility of tautomerism [
12] and the presence of a multifarious framework, offering versatility for applications in synthetic organic chemistry [
13,
14]. Derivatives of 3- or 5-amino pyrazole, generally referred to as 3(5)-aminopyrazoles, are especially interesting in this context, serving as starting materials for the synthesis of condensed heterocycles, including pyrazolo[1,5-a]pyrimidines, another privileged motif that attracted interest for many years and is extensively reviewed, in the past and also in recent literature [
15,
16,
17,
18]. However, to this date, a thorough investigation regarding the chemistry and diverse reactivity of 3(5)-aminopyrazoles leading to more complex heterocyclic systems has not been performed. A deeper understanding of the structure and chemistry of 3(5)-aminopyrazoles and pyrazoles in general would be beneficial for the elucidation of their chemical potential, their behavior in different environments and how their versatility may affect the development of efficient synthetic methodologies where pyrazoles feature. Several reviews on the chemistry of pyrazoles have been published [
19,
20,
21,
22,
23,
24,
25,
26,
27,
28,
29,
30,
31,
32,
33].
2. Pyrazole Properties
Pyrazole has a five-membered aromatic ring structure containing two vicinal nitrogen atoms, an acidic pyrrole-like nitrogen with a lone pair of electrons involved in aromaticity, a basic sp
2-hybridized pyridine-like nitrogen and three carbon atoms (
Figure 2) [
34], and these combined features must be carefully taken into account in the context of reactivity. In the first place, given the nature of the nitrogen, N-unsubstituted pyrazoles hold amphoteric properties, acting as both acids and bases. While the acidic pyrrole-like NH group easily donates its proton, the basic pyridine-like nitrogen has the ability to accept protons even more readily, and hence, the basic character is generally prevalent. Nevertheless, substitutions on the ring can modulate these properties, as, for instance, electron donating groups were shown to increase the acidity of the pyrrole-like NH group [
35,
36,
37]. Substituent effect and other modulators of proton transfer will be discussed in detail further in this work.
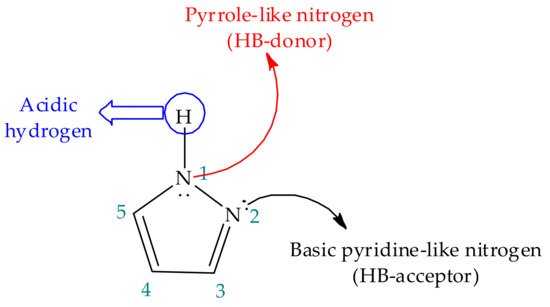
Figure 2. Representation of pyrazole structure, emphasizing the properties of the ring nitrogen atoms.
In addition to the previous, the combination of two dissimilar and adjacent nitrogen atoms in this azole (-N-N(H)- motif) allows it to simultaneously donate and accept hydrogen bonds (HB) (
Figure 3), which favors the establishment of intermolecular interactions, either among pyrazole molecules themselves, forming different types of linear and/or cyclic complexes contingent upon the physical state and the nature of the substituents in the ring, or between pyrazoles and neighboring molecules that participate in proton transfer processes [
38,
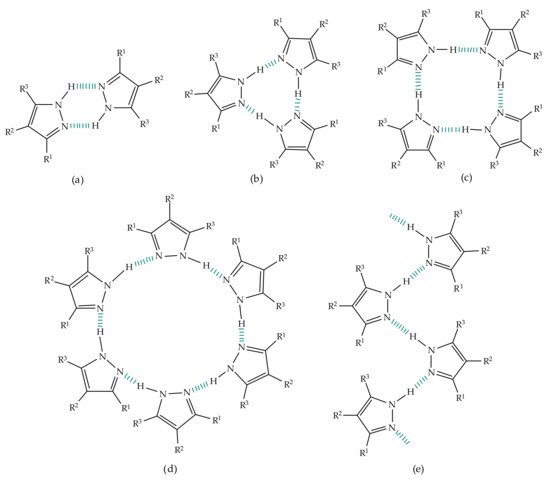
39]. Regarding the aggregation pattern of pyrazole in the solid state, X-ray crystal studies unraveled the formation of linear catemers as well as of cyclic dimers, trimers, tetramers and hexamers (
Figure 3) [
40,
41]. In solution, both linear and cyclic oligomers can form, but in this case the associations between pyrazole molecules depend strongly on the type of solvent, since more polar protic solvents can divert the intermolecular interactions towards themselves, favoring the pyrazole-solvent hydrogen bonding rather than formation of pyrazole-pyrazole clusters [
38,
40]. In the gas-phase, an intermolecular interaction also needs to take place to allow for proton transfer, whether it occurs with another pyrazole molecule or with a third molecule, or even results from collisions with the analytical instrument’s walls [
39,
42]. Pyrazole-based self-aggregates in the gas-phase have been detected by Infrared (IR) spectroscopy, for the parent pyrazole and for 3,5-dimethylpyrazole, as an equilibrium between monomers, dimers and trimmers [
38]. In addition, several theoretical studies were performed regarding intermolecular interactions in pyrazoles, leading to proton transfers in the gas phase [
38,
43,
44].
Figure 3. Representation of structures formed from self-association of pyrazole derivatives: (
a) dimers, (
b) trimers, (
c) tetramers, (
d) hexamers, (
e) catemers [
38,
39,
40,
41].
3. Tautomerism in Pyrazoles
Tautomerism is a key feature that stems from the aforementioned ability of pyrazoles to exchange protons. Ludwig Knorr, who discovered the mononuclear pyrazole motif as he stumbled upon a pyrazolone scaffold, during attempts to synthesize quinolones, was also one of the first chemists to contribute to the elucidation of the tautomerism phenomenon, back in the 19th century [
1,
45,
46].
Classically, the concept of tautomerism is associated to a mobile equilibrium between two or more isomeric structures that interconvert easily, with energetic barriers for interconversion below circa 20 kcal/mol, i.e., the different forms may coexist in the same medium [
47]. This phenomenon can manifest through several distinct forms, according to the type of exchange present. In heterocyclic systems, two criteria must be considered when discussing tautomerism: the structural aspect of the exchange and the nature of the exchanged element. Regarding the structural aspect, tautomerism is divided into four main types: annular tautomerism, side-chain tautomerism, ring-chain tautomerism and valence tautomerism. In the first type, as the name suggests, the alterations occur within the ring system, between annular carbon and heteroatoms such as nitrogen and oxygen; in the second type, the side-chain elements participate in the interconversion involving the ring; the other two types are characterized by transformations leading to bond formation or rupture—while in ring-chain tautomerism, the migration of the species on the side chain results in ring closure, the valence tautomerism is not a result of migrations of any group or element but rather a rupture or formation of bonds carried by an energetic stimulus [
47,
48,
49,
50]. Regarding the type of element involved in the interconversion, tautomerism can be subdivided into prototropy, when the exchange is characterized by the displacement of a proton between two positions suffering alteration, elementotropy, when a heavier element participates in the transformation, frequently a metal atom (metallotropy), and aniontropy and cationtropy, where the two isomers differ only on the position of an anion or cation, respectively [
42,
49,
51,
52].
Tautomeric equilibria do not happen solely within one molecule, they can be both intra and intermolecular phenomena. To set straight which kind of process is ongoing, one must take into account the accordance with the thermodynamic and kinetic principles of tautomerism as well as the substitution patterns and the environment conditions to which the compounds are submitted, such as the solvent medium, if present, since solvents usually catalyze isomeric transformations, as the presence and ratio of the tautomers strongly depend on these factors [
53]. In other words, many internal and external factors influence tautomeric equilibria, with the dielectric constant of the medium and the temperature also intervening as active modulators of these transformations [
47]. It was shown by Alkorta et al. that the proton exchange in pyrazoles is an intermolecular process rather than an intramolecular one, since the energy barrier determined for the intramolecular process displays values surrounding 50 kcal/mol, while the observed values for the intermolecular counterpart do not surpass the range of 10–14 kcal/mol [
54]. The information available indicates that protons migrate either with the help of third parties, such as solvent molecules, as for example water molecules were shown to reduce the activation energies when attached to pyrazole, both in the gas-phase and in liquid environment [
44], or of other pyrazole molecules, in self-assembled complexes.
4. Pyrazoles in Organic Synthesis
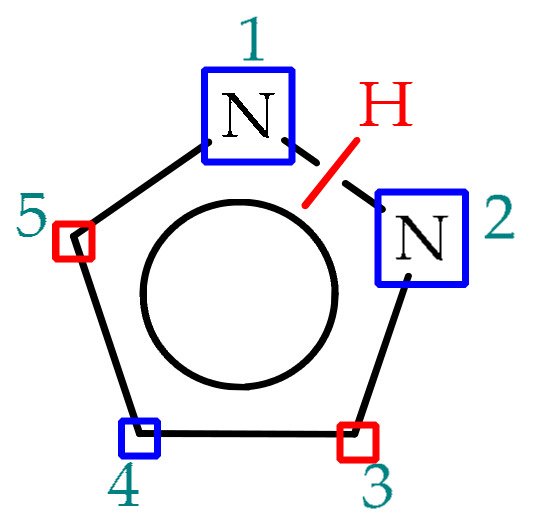
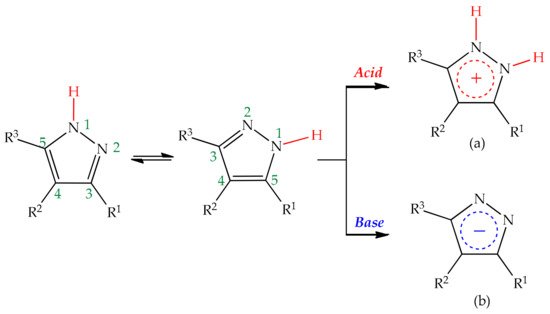
Pyrazoles are electron-rich heterocyclic systems which can be readily employed in organic synthesis, given their versatile chemistry. Within the pyrazole scaffold, besides the acidic and basic properties referred in the previous chapters, three positions display nucleophilic nature (N1, N2, C4) and two electrophilic ones (C3, C5), represented in
Figure 4 as blue and red rectangles, respectively. This way, pyrazole functionalization may be sought at any ring position, with nucleophiles adding preferentially to C3 and C5 positions, whereas electrophile addition occurs preferentially at C4 position and/or to any of the nitrogen atoms, depending on the reaction conditions. One way to efficiently introduce substituents at annular carbons is through directed cross-coupling reactions, such as Suzuki-couplings [
103], in which case C4-halogenations are usually necessary as prior steps to boronic acid or ester cross-coupling at the indicated position [
104].
Figure 4. Pyrazole structure highlighting the nucleophilic and electrophilic positions in the blue and red boxes, respectively.
It was previously stated that, in the presence of two annular nitrogen atoms with pyrrole and pyridine-like characters, the one participating in nucleophilic substitution is the basic pyridine-like nitrogen [
6]. However, it was later shown by Huang et al. that regioselective synthesis of N1 substituted pyrazoles could be achieved under specific conditions, which led them to investigate the electron density on the ring-nitrogen theoretically through density functional theory (DFT) calculations at the B3LYP/6-31G**(d) level, having concluded that the major negative charge was concentrated on the pyrrole-like nitrogen [
83]. Nevertheless, organic synthesis based on pyrazole synthons should regard carefully two main aspects that strongly influence the reactivity of the molecule, the medium or environment in which reactions occur and the substitution pattern on the ring system. On the one hand, the amphoteric nature of pyrazoles makes them especially propense to protonation or deprotonation reactions in acidic and basic environments, respectively, which alters considerably the nucleophilicity of the ring by originating a positively or negatively charged pyrazole ring (
Scheme 1).
Scheme 1. Schematic representation of the chemical behavior of pyrazoles in acid and basic media leading to pyrazole cations (a) and anions (b), respectively.
The nature of substituents, namely their size and electronic contributions, may also play a role in regiochemistry, altering the overall reactivity of the system. Kost stated that electron-withdrawing groups on the pyrazole ring increase its basicity, by increasing the acidity of the proton [
105]. Conversely, recent studies in 3(5)-substituted pyrazoles point to the opposite effect, with electron-donating groups at C3 increasing the basicity of the pyrazole ring [
37], as shown by theoretical calculations for 3(5)-methylpyrazole [
36]. Easy proton abstraction by bases generates the nucleophilic pyrazole anion (structure (b),
Scheme 1), which is prone to coupling with electrophilic moieties, wherein steric constraints imposed by substituents at C3 and C5 are usually the factors determining regioselectivity [
106]. Contrariwise, protonation of the pyridine-like nitrogen is also predisposed in acidic media, which in turn may direct the condensation reaction towards the participation of the exocyclic groups, as frequently happens in cyclization reactions involving 3(5)-aminopyrazoles [
13,
107]. Given that pyrazoles are tautomeric structures, protonation/deprotonation of the core structure could be one way to circumvent this issue, accounting for solvent and substituent properties, which often modulate reactivity and determine reaction pathways.
The versatility of the pyrazole motif offers a plethora of synthetic routes, enabling the preparation of libraries of compounds of interest to a broad community of organic and medicinal chemists. Synthetic methodologies should be carefully selected, and conditions should be optimized to favor regioselective processes. The literature continuously furnishes updates in synthetic methodologies involving pyrazoles and this topic has been covered in depth by several authors [
8,
19,
25,
26,
27,
108], including the meticulous and extensive review written by Fustero et al. [
33].
5. 3(5)-Aminopyrazole
5.1. Tautomerism in 3(5)-Aminopyrazole
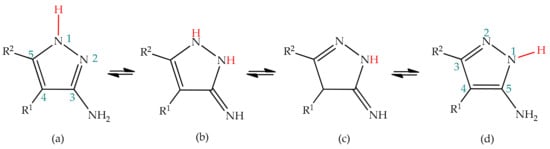
3(5)-Aminopyrazoles bear an amine substituent at the positions 3 or 5 of the pyrazole ring, as a result of prototropic annular tautomerism. In theory, the frame of 3(5)-aminopyrazole is also susceptible to side-chain tautomerism, given the presence of an amine side-chain prone to participate in proton exchange. Therefore, four tautomeric structures should be expected for 3(5)-aminopyrazoles, as depicted in
Scheme 2. However, studies indicate that the imino forms are not formed, and therefore only an annular rearrangement between positions 1 and 2 is predicted for 3(5)-aminopyrazoles (structures (a) and (d) of
Scheme 2), as happens for pyrazole [
50,
84].
Scheme 2. Side-chain tautomerism for 3(5)-aminopyrazole. The equilibrium between structures (a) and (d) represents the prototropic annular tautomerism for 3(5)-aminopyrazoles.
Tautomerism in 3(5)-aminopyrazoles has to be carefully appraised by chemists, on account of the specificities inherent to this scaffold that impact in reactivity and synthesis. Several experimental [
50,
72,
93,
109,
110,
111] and theoretical studies [
43,
60,
64,
70,
72,
84] were carried out, the majority focusing on the structural aspects and corroborating the general conclusions applicable to the pyrazole family. Nonetheless, studies clarifying the chemistry of aminopyrazoles as reactants in organic synthesis are scarce, especially on the unsubstituted form. Further efforts are needed to enlighten the scientific community on this matter.
5.2. 3(5)-Aminopyrazole in Heterocyclic Synthesis
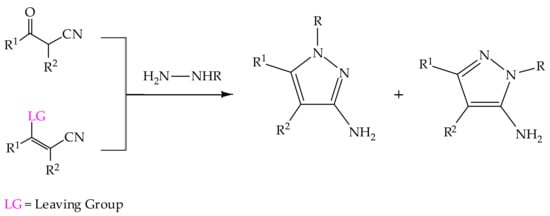
Synthesis of 3(5)-aminopyrazoles is usually carried out by incorporating the NH
2 group in the synthons rather than functionalization of the pre-built pyrazole, which corresponds most commonly to condensation reactions of 1,3-dielectrophilic nitriles and hydrazines, since this pathway represents the most efficient and straightforward strategy (
Scheme 3) [
114].
Scheme 3. General routes to the formation of the 3(5)-aminopyrazoles from 1,3-dielectrophilic nitriles and hydrazines [
20,
114].
The use of 3(5)-aminopyrazoles as building blocks in heterocyclic synthesis is challenging, mostly because these are polyfunctional compounds exhibiting a high chemical variability within the structure. 3(5)-aminopyrazoles are composed of one electrophilic C3/C5 position, depending on the considered tautomer, and four main nucleophilic sites: the two ring nitrogen atoms, the 4-carbon and the exocyclic amine. These characteristics must be duly considered in the design of synthetic methodologies, since for example the nucleophilic positions display distinct reactivities, i.e., while the C4 carbon participates in electrophilic aromatic substitutions, the nitrogen atoms mediate typical S
N2 reactions. These properties have been widely explored in the synthesis of fused heterocycles [
107], which is of our particular interest, in view of the construction of pyrazolo[1,5-a]pyrimidines.
Recent reviews cover synthetic methodologies using 3(5)-aminopyrazoles as buildings blocks to the present state of the art [
13,
16,
107]. However, the majority of these works fail to explain the discrepancies noted in somewhat similar conditions and reactivity aspects linked to the tautomeric precursor are elusive. For this reason, our focus in this review is on the chemical potential of 3(5)-aminopyrazoles in heterocyclic synthesis. As stated by El-Sattar et al., the chemical potential of a compound reflects the relationship between its structure and reactivity, thus a comparatively higher chemical potential of a compound means a higher reactivity [
115]. In compounds that exhibit tautomerism, the reactivity may be controlled by a particular tautomeric form and, according to the Gustafsson paradox, the less abundant tautomer tends to be the most reactive one [
116]. In these circumstances, a paramount need to elucidate the precise chemical potential of the entities present in a specific reactant mixture emerges. HOMO and LUMO states and energies of the tautomeric structures represent the parameters that mostly contribute to their chemical potential; as such, variations alter the acidic/basic and electrophilic/nucleophilic properties of the compounds [
85,
115]. It was already shown that in heterocycles, including 3(5)-aminopyrazoles, the different characteristics of molecular orbitals promote distinct reactivities of each of the tautomeric species [
66,
85]. For 3(5)-aminopyrazole, from the information gathered in the present work, and given the nature of the NH
2 substituent, one may infer that the 3-tautomer should be the most stable one. Conversely, the majority of synthetic strategies to produce more complex heterocyclic systems rely on the 5-tautomer to be pursued.