 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Isidro Ferrer | -- | 2175 | 2022-06-29 12:34:49 | | | |
| 2 | Vivi Li | Meta information modification | 2175 | 2022-06-30 08:36:25 | | |
Video Upload Options
Primary microglial leukodystrophy or leukoencephalopathy are disorders in which a genetic defect linked to microglia causes cerebral white matter damage. Pigmented orthochromatic leukodystrophy, adult-onset orthochromatic leukodystrophy associated with pigmented macrophages, hereditary diffuse leukoencephalopathy with (axonal) spheroids, and adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) are different terms apparently used to designate the same disease. However, ALSP linked to dominantly inherited mutations in CSF1R (colony-stimulating factor receptor 1) causes CSF-1R-related leukoencephalopathy (CRP). Yet, recessive ALSP with ovarian failure linked to AARS2 (alanyl-transfer (t)RNA synthase 2) mutations (LKENP) is a mitochondrial disease and not a primary microglial leukoencephalopathy. Polycystic membranous lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL; Nasu–Hakola disease: NHD) is a systemic disease affecting bones, cerebral white matter, selected grey nuclei, and adipose tissue The disease is caused by mutations of one of the two genes TYROBP or TREM2, identified as PLOSL1 and PLOSL2, respectively.
1. Introduction
2. Adult-Onset Leukodystrophy with Axonal Spheroids and Pigmented Glia (POLD, HDLS, ALSP): CRP Linked to Mutations in CSF1R, and LKENP (Leukoencephalopathy, Progressive with Ovarian Failure) Linked to Heterozygous Mutations in AARS2
2.1. Clinical Features
2.1.1. ALSP-CSFR1 or CRL
2.1.2. ALSP-AARS2 or LKENP
2.2. Radiological Findings
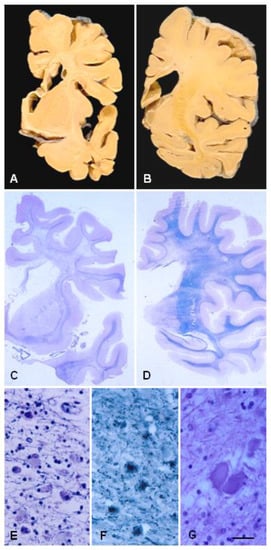
2.3. Neuropathology
3. Polycystic Membranous Lipomembranous Osteodysplasia with Sclerosing Leukoencephalopathy (PLOSL; Nasu–Hakola Disease)
3.1. Clinical Features
3.2. Radiological Examination
3.3. General Pathology
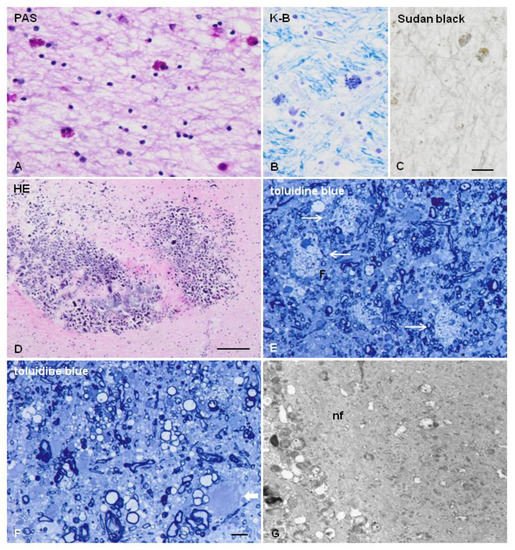
3.4. Neuropathology
References
- Van Der Knaap, M.S.; Bugiani, M. Leukodystrophies: A proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol. 2017, 134, 351–382.
- Köhler, W.; Curiel, J.; Vanderver, A. Adulthood leukodystrophies. Nat. Rev. Neurol. 2018, 14, 94–105.
- Lynch, D.S.; De Paiva, A.R.B.; Zhang, W.; Bugiardini, E.; Freua, F.; Lucato, L.; de Souza, L.I.M.; Lakshmanan, R.; Kinsella, J.A.; Merwick, A.; et al. Clinical and genetic characterization of leukoencephalopathies in adults. Brain 2017, 140, 1204–1211.
- Van der Knaap, M.S.; Schiffmann, R.; Mochel, F.; Wolf, N. Diagnosis, prognosis, and treatment of leukodystrophies. Lancet Neurol. 2019, 18, 962–972.
- Resende, L.L.; De Paiva, A.R.B.; Kok, F.; Leite, C.D.C.; Lucato, L. Adult Leukodystrophies: A Step-by-Step Diagnostic Approach. RadioGraphics 2019, 39, 153–168.
- Garcia, L.M.; Hacker, J.L.; Sase, S.; Adang, L.; Almad, A. Glial cells in the driver seat of leukodystrophy pathogenesis. Neurobiol. Dis. 2020, 146, 105087.
- Konno, T.; Yoshida, K.; Mizuno, T.; Kawarai, T.; Tada, M.; Nozaki, H.; Ikeda, S.-I.; Nishizawa, M.; Onodera, O.; Wszolek, Z.K.; et al. Clinical and genetic characterization of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia associated with CSF1R mutation. Eur. J. Neurol. 2016, 24, 37–45.
- Freeman, S.H.; Hyman, B.T.; Sims, K.B.; Hedley-Whyte, E.T.; Vossough, A.; Frosch, M.P.; Schmahmann, J.D. Adult Onset Leukodystrophy with Neuroaxonal Spheroids: Clinical, Neuroimaging and Neuropathologic Observations. Brain Pathol. 2008, 19, 39–47.
- Lynch, D.S.; Jaunmuktane, Z.; Sheerin, U.-M.; Phadke, R.; Brandner, S.; Milonas, I.; Dean, A.; Bajaj, N.; McNicholas, N.; Costello, D.; et al. Hereditary leukoencephalopathy with axonal spheroids: A spectrum of phenotypes from CNS vasculitis to parkinsonism in an adult onset leukodystrophy series. J. Neurol. Neurosurg. Psychiatry 2015, 87, 512–519.
- Sundal, C.; Fujioka, S.; Van Gerpen, J.A.; Wider, C.; Nicholson, A.M.; Baker, M.; Shuster, E.A.; Aasly, J.; Spina, S.; Ghetti, B.; et al. Parkinsonian features in hereditary diffuse leukoencephalopathy with spheroids (HDLS) and CSF1R mutations. Park. Relat. Disord. 2013, 19, 869–877.
- Kleinfeld, K.; Mobley, B.; Hedera, P.; Wegner, A.; Sriram, S.; Pawate, S. Adult-onset leukoencephalopathy with neuroaxonal spheroids and pigmented glia: Report of five cases and a new mutation. J. Neurol. 2012, 260, 558–571.
- Kimura, T.; Ishizawa, K.; Mitsufuji, T.; Abe, T.; Nakazato, Y.; Yoshida, K.; Sasaki, A.; Araki, N. A clinicopathological and genetic study of sporadic diffuse leukoencephalopathy with spheroids: A report of two cases. Neuropathol. Appl. Neurobiol. 2013, 39, 837–843.
- Guerreiro, R.; Kara, E.; Le Ber, I.; Bras, J.; Rohrer, J.D.; Taipa, R.; Lashley, T.; Dupuits, C.; Gurunlian, N.; Mochel, F.; et al. Genetic Analysis of Inherited Leukodystrophies. JAMA Neurol. 2013, 70, 875–882.
- Konno, T.; Tada, M.; Koyama, A.; Nozaki, H.; Harigaya, Y.; Nishimiya, J.; Matsunaga, A.; Yoshikura, N.; Ishihara, K.; Arakawa, M.; et al. Haploinsufficiency of CSF-1R and clinicopathologic characterization in patients with HDLS. Neurology 2013, 82, 139–148.
- Kawakami, I.; Iseki, E.; Kasanuki, K.; Minegishi, M.; Sato, K.; Hino, H.; Shibuya, K.; Fujisawa, K.; Higashi, S.; Akiyama, H.; et al. A family with hereditary diffuse leukoencephalopathy with spheroids caused by a novel c.2442+2T>C mutation in the CSF1R gene. J. Neurol. Sci. 2016, 367, 349–355.
- Ikeuchi, T.; Mezaki, N.; Miura, T. Cognitive dysfunction and symptoms of movement disorders in adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. Park. Relat. Disord. 2018, 46, S39–S41.
- Leng, C.; Lu, L.; Wang, G.; Zhang, Y.; Xu, Y.; Lin, X.; Shen, N.; Xu, X.; Qun, S.; Sun, M.; et al. A novel dominant-negative mutation of the CSF1R gene causes adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. Am. J. Transl. Res. 2019, 11, 6093–6101.
- Sundal, C.; Wszolek, Z.K. CSFR1-related adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. In GeneReviews® ; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2017.
- Calatayud, T.; Turkalp, Z.T.; Gonzales, A.A.; Muñoz, D.G. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: Report on a case with morphometric studies. Clin. Neuropathol. 2013, 32, 492–501.
- Tian, W.-T.; Zhan, F.-X.; Liu, Q.; Luan, X.-H.; Zhang, C.; Shang, L.; Zhang, B.-Y.; Pan, S.-J.; Miao, F.; Hu, J.; et al. Clinicopathologic characterization and abnormal autophagy of CSF1R-related leukoencephalopathy. Transl. Neurodegener. 2019, 8, 32.
- Funayama, M.; Sugihara, M.; Takata, T.; Mimura, M.; Ikeuchi, T. Remarkable behavioural signs and progressive non-fluent aphasia in a patient with adult-onset leucoencephalopathy with axonal spheroids and pigmented glia. Psychogeriatrics 2018, 19, 282–285.
- Di Donato, I.; Stabile, C.; Bianchi, S.; Taglia, I.; Mignarri, A.; Salvatore, S.; Giorgio, E.; Brusco, A.; Simone, I.; Dotti, M.T.; et al. A Novel CSF1R Mutation in a Patient with Clinical and Neuroradiological Features of Hereditary Diffuse Leukoencephalopathy with Axonal Spheroids. J. Alzheimer’s Dis. 2015, 47, 319–322.
- Shu, Y.; Long, L.; Liao, S.; Yang, J.; Li, J.; Qiu, W.; Yang, Y.; Bao, J.; Wu, A.; Hu, X.; et al. Involvement of the optic nerve in mutated CSF1R-induced hereditary diffuse leukoencephalopathy with axonal spheroids. BMC Neurol. 2016, 16, 171.
- Lynch, D.S.; Zhang, W.; Lakshmanan, R.; Kinsella, J.A.; Uzun, G.A.; Karbay, M.; Tufekcioglu, Z.; Hanagasi, H.; Burke, G.; Foulds, N.; et al. Analysis of Mutations in AARS2 in a Series of CSF1R-Negative Patients with Adult-Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia. JAMA Neurol. 2016, 73, 1433–1439.
- Wang, M.; Zhang, X. A novel CSF-1R mutation in a family with hereditary diffuse leukoencephalopathy with axonal spheroids misdiagnosed as hydrocephalus. Neurogenetics 2019, 20, 155–160.
- Mao, C.; Zhou, L.; Zhou, L.; Yang, Y.; Niu, J.; Li, J.; Huang, X.; Ren, H.; Zhao, Y.; Peng, B.; et al. Biopsy histopathology in the diagnosis of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP). Neurol. Sci. 2019, 41, 403–409.
- Dallabona, C.; Diodato, D.; Kevelam, S.H.; Haack, T.B.; Wong, L.-J.; Salomons, G.S.; Baruffini, E.; Melchionda, L.; Mariotti, C.; Strom, T.M.; et al. Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology 2014, 82, 2063–2071.
- Dong, Q.; Long, L.; Chang, Y.-Y.; Lin, Y.-J.; Liu, M.; Lu, Z.-Q. An adolescence-onset male leukoencephalopathy with remarkable cerebellar atrophy and novel compound heterozygous AARS2 gene mutations: A case report. J. Hum. Genet. 2018, 63, 841–846.
- Itoh, K.; Shiga, K.; Shimizu, K.; Muranishi, M.; Nakagawa, M.; Fushiki, S. Autosomal dominant leukodystrophy with axonal spheroids and pigmented glia: Clinical and neuropathological characteristics. Acta Neuropathol. 2005, 111, 39–45.
- Van Gerpen, J.A.; Wider, C.; Broderick, D.F.; Dickson, D.W.; Brown, L.A.; Wszolek, Z.K. Insights into the dynamics of hereditary diffuse leukoencephalopathy with axonal spheroids. Neurology 2008, 71, 925–929.
- Sundal, C.; Van Gerpen, J.A.; Nicholson, A.M.; Wider, C.; Shuster, E.A.; Aasly, J.; Spina, S.; Ghetti, B.; Roeber, S.; Garbern, J.; et al. MRI characteristics and scoring in HDLS due to CSF1R gene mutations. Neurology 2012, 79, 566–574.
- Fujioka, S.; Broderick, D.F.; Sundal, C.; Baker, M.C.; Rademakers, R.; Wszolek, Z.K. An adult-onset leukoencephalopathy with axonal spheroids and pigmented glia accompanied by brain calcifications: A case report and a literature review of brain calcifications disorders. J. Neurol. 2013, 260, 2665–2668.
- Bender, B.; Klose, U.; Lindig, T.; Biskup, S.; Nägele, T.; Schöls, L.; Karle, K.N. Imaging features in conventional MRI, spectroscopy and diffusion weighted images of hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS). J. Neurol. 2014, 261, 2351–2359.
- Robinson, J.L.; Suh, E.; Wood, E.M.; Lee, E.B.; Coslett, H.B.; Raible, K.; Lee, V.M.-Y.; Trojanowski, J.Q.; Van Deerlin, V.M. Common neuropathological features underlie distinct clinical presentations in three siblings with hereditary diffuse leukoencephalopathy with spheroids caused by CSF1R p.Arg782His. Acta Neuropathol. Commun. 2015, 3, 42.
- Ayrignac, X.; Nicolas, G.; Carra-Dallière, C.; Hannequin, D.; Labauge, P. Brain Calcifications in Adult-Onset Genetic Leukoencephalopathies. JAMA Neurol. 2017, 74, 1000.
- Codjia, P.; Ayrignac, X.; Mochel, F.; Mouzat, K.; Carra-Dalliere, C.; Castelnovo, G.; Ellie, E.; Etcharry-Bouyx, F.; Verny, C.; Belliard, S.; et al. Adult-Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia: An MRI Study of 16 French Cases. Am. J. Neuroradiol. 2018, 39, 1657–1661.
- Konno, T.; Broderick, D.F.; Mezaki, N.; Isami, A.; Kaneda, D.; Tashiro, Y.; Tokutake, T.; Keegan, B.M.; Woodruff, B.K.; Miura, T.; et al. Diagnostic Value of Brain Calcifications in Adult-Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia. Am. J. Neuroradiol. 2016, 38, 77–83.
- Abe, T.; Kawarai, T.; Fujita, K.; Sako, W.; Terasawa, Y.; Matsuda, T.; Sakai, W.; Tsukamoto-Miyashiro, A.; Matsui, N.; Izumi, Y.; et al. MR Spectroscopy in Patients with Hereditary Diffuse Leukoencephalopathy with Spheroids and Asymptomatic Carriers of Colony-stimulating Factor 1 Receptor Mutation. Magn. Reson. Med Sci. 2017, 16, 297–303.
- Gray, F.; Destée, A.; Bourre, J.-M.; Gherardi, R.; Krivosic, I.; Warot, P.; Poirier, J. Pigmentary Type of Orthochromatic Leukodystrophy: A new case with ultrastructural and biochemical study. J. Neuropathol. Exp. Neurol. 1987, 46, 585–596.
- Tuñón, T.; Ferrer, I.; Gállego, J.; Delgado, G.; Villanueva, J.A.; Martinez-Peñuela, J.M. Leucodystrophy with pigmented glial and scavenger cells (pigmentary type of orthochromatic leucodystrophy). Neuropathol. Appl. Neurobiol. 1988, 14, 337–344.
- Hoffmann, S.; Murrell, J.; Harms, L.; Miller, K.; Meisel, A.; Brosch, T.; Scheel, M.; Ghetti, B.; Goebel, H.-H.; Stenzel, W. Enlarging the Nosological Spectrum of Hereditary Diffuse Leukoencephalopathy with Axonal Spheroids (HDLS). Brain Pathol. 2014, 24, 452–458.
- Mendes, A.; Pinto, M.; Vieira, S.; Castro, L.; Carpenter, S. Adult-onset leukodystrophy with axonal spheroids. J. Neurol. Sci. 2010, 297, 40–45.
- Ali, Z.S.; Van Der Voorn, J.P.; Powers, J.M. A Comparative Morphologic Analysis of Adult Onset Leukodystrophy with Neuroaxonal Spheroids and Pigmented Glia-A Role for Oxidative Damage. J. Neuropathol. Exp. Neurol. 2007, 66, 660–672.
- Martinez-Saez, E.; Shah, S.; Costa, C.; Fleminger, S.; Connor, S.; Bodi, I. Adult onset leukodystrophy with neuroaxonal spheroids and demyelinating plaque-like lesions. Neuropathology 2011, 32, 285–292.
- Alturkustani, M.; Sharma, M.; Hammond, R.; Ang, L.-C. Adult-Onset Leukodystrophy: Review of 3 Clinicopathologic Phenotypes and a Proposed Classification. J. Neuropathol. Exp. Neurol. 2013, 72, 1090–1103.
- Oyanagi, K.; Kinoshita, M.; Inoue, T.; Nakahara, A.; Tokiwai, M.; Arai, N.; Aoki, N.; Jinnai, K.; Yazawa, I.; Ishihara, K.; et al. Adult onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) and Nasu-Hakola disease: Lesion staging and dynamic changes of axons and microglial subsets. Brain Pathol. 2017, 27, 748–769.
- Alturkustani, M.; Keith, J.; Hazrati, L.-N.; Rademakers, R.; Ang, L.-C. Pathologic Staging of White Matter Lesions in Adult-Onset Leukoencephalopathy/Leukodystrophy With Axonal Spheroids. J. Neuropathol. Exp. Neurol. 2015, 74, 233–240.
- Lin, W.L.; Wszolek, Z.K.; Dickson, D.W. Hereditary diffuse leukoencephalopathy with spheroids: Ultrastructural and immunoe-lectron microscopic studies. Int. J. Clin. Exp. Pathol. 2010, 3, 665–674.
- Terayama, K. Two cases of cystic bone disease showing peculiar features. Nippon Seikeigeka Gakkai Zasshi 1961, 35, 626.
- Hakola, H.P.A.; Järvi, O.H.; Sourander, P. Osteodysplasia polycystica hereditaria combined with sclerosing leucoencephalopathy. Acta Neurol. Scand. 2009, 46, 79–80.
- Nasu, T.; Tsukahara, Y.; Terayama, K. A lipid metabolic disease—“Membranous lipodystrophy”—An autopsy case demonstrating numerous peculiar membrane-structures composed of compound lipid in bone and bone marrow and various adipose tissues. Acta Pathol. Jpn. 1973, 23, 539–558.
- Harada, K. A case of “membranous lipodystrophy (Nasu)” with emphasis on psychiatric and neuropathologic aspects. Folia Psychiatr. Neurol. Jpn. 1975, 29, 169–177.
- Hakola, P.; Virtama, P. Radiologic bone changes of polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy. Skelet. Radiol. 1982, 8, 51–54.
- Minagawa, M.; Maeshiro, H.; Shioda, K.; Hirano, A. Membranous lipodystrophy (Nasu disease): Clinical and neuropathological study of a case. Clin. Neuropathol. 1985, 4, 38–45.
- Hakola, H.P. Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (membranous lipodystrophy): A neuropsychiatric follow-up study. In Monographs of Psychiatria Fennica; Henriksson, M., Huttunen, M., Kuoppasalmi, K., Lindfors, O., Lonnqvist, J., Eds.; Foundation for Psychiatric Research: Helsinki, Finland, 1990; pp. 1–114.
- Ishigooka, M.; Hashimoto, T.; Izumiya, K.; Kodama, C.; Nakada, T. Membranous Lipodystrophy (Nasu’s Disease): A Rare Cause of Neuropathic Urinary Incontinence. Urol. Int. 1993, 50, 179–181.
- Verloes, A.; Maquet, P.; Sadzot, B.; Vivario, M.; Thiry, A.; Franck, G. Nasu-Hakola syndrome: Polycystic lipomembranous osteodysplasia with sclerosing leucoencephalopathy and presenile dementia. J. Med. Genet. 1997, 34, 753–757.
- Bm, J.P.; Autti, T.; Raininko, R.; Partanen, J.; Salonen, O.; Puranen, M.; Hakola, P.; Haltia, M. CNS manifestations of Nasu-Hakola disease: A frontal dementia with bone cysts. Neurology 2001, 56, 1552–1558.
- Bianchin, M.M.; Capella, H.M.; Chaves, D.L.; Steindel, M.; Grisard, E.C.; Ganev, G.G.; da Silva Júnior, J.P.; Neto Evaldo, S.; Poffo, M.A.; Walz, R.; et al. Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy—PLOSL): A dementia associated with bone cystic lesions. From clinical to genetic and molecular aspects. Cell Mol. Neurobiol. 2004, 24, 1–24.
- Kaneko, M.; Sano, K.; Nakayama, J.; Amano, N. Nasu-Hakola disease: The first case reported by Nasu and review: The 50th An-niversary of Japanese Society of Neuropathology. Neuropathology 2010, 30, 463–470.
- Ilonen, T.; Hakola, P.; Vanhanen, M.; Tiihonen, J. Rorschach assessment of personality functioning in patients with polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy. Acta Neuropsychiatr. 2012, 24, 236–244.
- Paloneva, J.; Autti, T.; Hakola, P.; Haltia, M.J. Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL). In GeneReviews® ; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2020.
- Kitajima, I.; Kuriyama, M.; Usuki, F.; Izumo, S.; Osame, M.; Suganuma, T.; Murata, F.; Nagamatsu, K. Nasu-Hakola disease (membranous lipodystrophy): Clinical, histopathological and biochemical studies of three cases. J. Neurol. Sci. 1989, 91, 35–52.
- Montalbetti, L.; Ratti, M.T.; Greco, B.; Aprile, C.; Moglia, A.; Soragna, D. Neuropsychological tests and functional nuclear neuroim-aging provide evidence of subclinical impairment in Nasu-Hakola disease heterozygotes. Funct. Neurol. 2005, 20, 71–75.
- Chouery, E.; Delague, V.; Bergougnoux, A.; Koussa, S.; Serre, J.-L.; Mégarbané, A. Mutations in TREM2 lead to pure early-onset dementia without bone cysts. Hum. Mutat. 2008, 29, E194–E204.
- Guerreiro, R.; Bilgic, B.; Guven, G.; Bras, J.; Rohrer, J.; Lohmann, E.; Hanagasi, H.; Gürvit, H.; Emre, M. A novel compound heterozygous mutation in TREM2 found in a Turkish frontotemporal dementia-like family. Neurobiol. Aging 2013, 34, 2890.e1–2890.e5.
- Guerreiro, R.J.; Lohmann, E.; Bras, J.; Gibbs, J.R.; Rohrer, J.; Gurunlian, N.; Dursun, B.; Bilgic, B.; Hanagasi, H.; Gürvit, H.; et al. Using Exome Sequencing to Reveal Mutations in TREM2 Presenting as a Frontotemporal Dementia–like Syndrome Without Bone Involvement. JAMA Neurol. 2013, 70, 78–84.
- Bock, V.; Botturi, A.; Gaviani, P.; Lamperti, E.; Maccagnano, C.; Piccio, L.; Silvani, A.; Salmaggi, A. Polycystic Lipomembranous Osteodysplasia with Sclerosing Leukoencephalopathy (PLOSL): A new report of an Italian woman and review of the literature. J. Neurol. Sci. 2013, 326, 115–119.
- Le Ber, I.; De Septenville, A.; Guerreiro, R.; Bras, J.; Camuzat, A.; Caroppo, P.; Lattante, S.; Couarch, P.; Kabashi, E.; Bouya-Ahmed, K.; et al. Homozygous TREM2 mutation in a family with atypical frontotemporal dementia. Neurobiol. Aging 2014, 35, 2419.e23–2419.e25.
- Matsuo, T.; Suetsugu, M.; Eguchi, M.; Sasaki, M.; Tsuneyoshi, M. Membranous lipodystrophy. A case report. Arch Psychiatr. Nervenkr. 1982, 231, 123–130.
- Hakola, H.P. Neuropsychiatric and genetic aspects of a new hereditary disease characterized by progressive dementia and lipomembranous polycystic osteodysplasia. Acta Psychiatr. Scand. Suppl. 1972, 232, 1–73.
- Nwawka, O.K.; Schneider, R.; Bansal, M.; Mintz, U.N.; Lane, J. Membranous lipodystrophy: Skeletal findings on CT and MRI. Skelet. Radiol. 2014, 43, 1449–1455.
- Hakola, H.P.; Partanen, V.S. Neurophysiological findings in the hereditary presenile dementia characterised by polycystic lipomembranous osteodysplasia and sclerosing leukoencephalopathy. J. Neurol. Neurosurg. Psychiatry 1983, 46, 515–520.
- Bird, T.D.; Koerker, R.M.; Leaird, B.J.; Vlcek, B.W.; Thorning, D.R. Lipomembranous polycystic osteodysplasia (brain, bone, and fat disease): A genetic cause of presenile dementia. Neurology 1983, 33, 81.
- Shibata, K.; Uchiyama, S.; Takeuchi, M.; Kobayashi, I.; Maruyama, S. A case of membranous lipodystrophy (Nasu) with diffuse cerebral, white matter involvement and cerebellar atrophy on brain CT and NM (in Japanese). Rinsho Shinkeigaku 1990, 30, 1232–1237.
- Araki, T.; Ohba, H.; Monzawa, S.; Sakuyama, K.; Hachiya, J.; Seki, T.; Takahashi, Y.; Yamaguchi, M. Membranous lipodystrophy: MR imaging appearance of the brain. Radiology 1991, 180, 793–797.
- Coomans, C.; Sieben, A.; Lammens, M.; Groote, C.C.-D.; Vandenbroecke, C.; Goethals, I.; Van Melkebeke, D.; Hemelsoet, D. Early-onset dementia, leukoencephalopathy and brain calcifications: A clinical, imaging and pathological comparison of ALSP and PLOSL/Nasu Hakola disease. Acta Neurol. Belg. 2018, 118, 607–615.
- Takeshita, T.; Kaminaga, T.; Tatsumi, T.; Hatanaka, Y.; Furui, S. Regional cerebral blood flow in a patient with Nasu-Hakola disease. Ann. Nucl. Med. 2005, 19, 309–312.
- Hakola, H.P.A.; Iivanainen, M. A new hereditary disease with progressive dementia and polycystic osteodysplasia: Neuroradiological analysis of seven cases. Neuroradiology 1973, 6, 162–168.
- Klunemann, H.H.; Ridha, B.H.; Magy, L.; Wherrett, J.R.; Hemelsoet, D.; Keen, R.W.; De Bleecker, J.L.; Rossor, M.; Marienhagen, J.; Klein, H.E.; et al. The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology 2005, 64, 1502–1507.
- Sageshima, M.; Masuda, H.; Kawamura, K.; Shozawa, T. Membranous lipodystrophy. Light and electron microscopic study of a biopsy case. Acta Pathol. Jpn. 1987, 37, 281–290.
- Machinami, R. Degenerative change of adipose tissue; the so-called membranous lipodystrophy. Virchows Arch. 1990, 416, 373–374.
- Mii, Y.; Miyauchi, Y.; Yoshikawa, T.; Honoki, K.; Aoki, M.; Tsutsumi, M.; Maruyama, H.; Funauchi, M.; Konishi, Y.; Tamai, S. Ultrastructural lipid and glycoconjugate cytochemistry of membranous lipodystrophy (Nasu-Hakola disease). Virchows Arch. 1991, 419, 137–142.
- Shboul, M.; Roschger, P.; Ganger, R.; Paschalis, L.; Rokidi, S.; Zandieh, S.; Behunova, J.; Muschitz, C.; Fahrleitner-Pammer, A.; Ng, A.Y.J.; et al. Bone matrix hypermineralization associated with low bone turnover in a case of Nasu-Hakola disease. Bone 2018, 123, 48–55.
- Yagishita, S.; Ito, Y.; Sakai, H.; Amano, N. Membranocystic lesions of the lung in Nasu-Hakola disease. Virchows Arch. 1985, 408, 211–217.
- Tanaka, J. Nasu-Hakola disease: A review of its leukoencephalopathic and membranolipodystrophic features. Neuropathology 2000, 20, 25–29.
- Aoki, N.; Tsuchiya, K.; Togo, T.; Kobayashi, Z.; Uchikado, H.; Katsuse, O.; Suzuki, K.; Fujishiro, H.; Arai, T.; Iseki, E.; et al. Gray matter lesions in Nasu-Hakola disease: A report on three autopsy cases. Neuropathology 2010, 31, 135–143.
- Kalimo, H.; Sourander, P.; Jarvi, O.; Hakola, P. Vascular changes and blood-brain barrier damage in the pathogenesis of polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (membranous lipodystrophy). Acta. Neurol. Scand. 1994, 89, 353–356.
- Matsushita, M.; Oyanagi, S.; Hanawa, S.; Shiraki, H.; Kosaka, K. Nasu-Hakola’s disease (membranous lipodystrophy). Acta Neuropathol. 1981, 54, 89–93.
- Miyazu, K.; Kobayashi, K.; Fukutani, Y.; Nakamura, I.; Hasegawa, H.; Yamaguchi, N.; Saitoh, T. Membranous lipodystrophy (Nasu-Hakola disease) with thalamic degeneration: Report of an autopsied case. Acta Neuropathol. 1991, 82, 414–419.
- Satoh, J.-I.; Kino, Y.; Yanaizu, M.; Saito, Y. Alzheimer’s disease pathology in Nasu-Hakola disease brains. Intractable Rare Dis. Res. 2018, 7, 32–36.
- Maderna, E.; Visonà, S.; Bolcato, V.; Redaelli, V.; Caroppo, P.; Montalbetti, L.; Giaccone, G.; Osculati, A. Neuropathological Alzheimer’s Disease Lesions in Nasu-Hakola Disease with TREM2 Mutation: Atypical Distribution of Neurofibrillary Changes. J. Alzheimer’s Dis. 2021, 79, 25–30.
- Yokoi, S.; Suzuki, K.; Amano, N.; Yagishita, S. Fatty Acid Analysis of Galactolipids and Ganglioside in the Brains of Four Cases of Nasu-Hakola Disease. Psychiatry Clin. Neurosci. 1989, 43, 695–701.
- Iannaccone, S.; Ferini-Strambi, L.; Nemni, R.; Marchettini, P.; Corbo, M.; Pinto, P.; Smirne, S. Pheripheral motor-sensory neuropathy in membranous lipodystrophy (Nasu’s disease): A case report. Clin. Neuropathol. 1992, 11, 49–53.

