Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Angel Ortega | -- | 2722 | 2022-06-27 08:09:24 | | | |
| 2 | Camila Xu | Meta information modification | 2722 | 2022-06-27 08:24:45 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Navarro, C.; Ortega, �.; Santeliz, R.; Garrido, B.; Chacín, M.; Galban, N.; Vera, I.; Sanctis, J.D.; Bermúdez, V. Metabolic Reprogramming in Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/24495 (accessed on 11 August 2026).
Navarro C, Ortega �, Santeliz R, Garrido B, Chacín M, Galban N, et al. Metabolic Reprogramming in Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/24495. Accessed August 11, 2026.
Navarro, Carla, Ángel Ortega, Raquel Santeliz, Bermary Garrido, Maricarmen Chacín, Néstor Galban, Ivana Vera, Juan De Sanctis, Valmore Bermúdez. "Metabolic Reprogramming in Cancer" Encyclopedia, https://encyclopedia.pub/entry/24495 (accessed August 11, 2026).
Navarro, C., Ortega, �., Santeliz, R., Garrido, B., Chacín, M., Galban, N., Vera, I., Sanctis, J.D., & Bermúdez, V. (2022, June 27). Metabolic Reprogramming in Cancer. In Encyclopedia. https://encyclopedia.pub/entry/24495
Navarro, Carla, et al. "Metabolic Reprogramming in Cancer." Encyclopedia. Web. 27 June, 2022.
Copy Citation
Reprogramming is a process mediated by multiple factors, including oncogenes, growth factors, hypoxia-induced factors, and the loss of suppressor gene function, which support malignant transformation and tumor development in addition to cell heterogeneity. Consequently, this hallmark promotes resistance to conventional anti-tumor therapies by adapting to the drastic changes in the nutrient microenvironment that these therapies entail. Therefore, it represents a revolutionary landscape during cancer progression that could be useful for developing new and improved therapeutic strategies targeting alterations in cancer cell metabolism, such as the deregulated mTOR and PI3K pathways.
metabolic reprogramming
tumor microenvironment
energy metabolism
1. Introduction
Cancer is a term used to define a group of diseases characterized by abnormal cell growth with an autonomous and uncontrolled expansion [1]. It represents a significant and escalating public health issue, responsible for one in six deaths worldwide. In this regard, in 2020, 19.3 million new cases and 10.0 million deaths from cancer were estimated [2], numbers that continue to increase, such that by the year 2040, cancer could be diagnosed in more than 29.4 million people per year [3].
It is currently known that cancer cells develop numerous properties that differentiate them from non-cancerous cells. These properties are known as hallmarks and include active proliferation, evasion of growth suppressors, resistance to cell death, angiogenesis, invasion and metastasis, immune evasion, tumour-promoting inflammation, genomic instability, mutation, non-mutational epigenetic reprogramming, polymorphic microbiomes, phenotypic plasticity, and metabolic reprogramming [3][4]. The latter refers to the ability of cancer cells to modify their metabolism, increasing the absorption and use of carbohydrates, lipids, and proteins to provide a pro-tumorigenic response in the face of the acquisition and maintenance of malignant properties [5].
Many factors mediate metabolic reprogramming: oncogenes, growth factors, hypoxia-induced factors, and the dysfunction of tumor suppressor genes. These alterations cause alterations in cell metabolism, especially glucose, whose absorption rate drastically increases in cancer [6].
Metabolic reprogramming allows cancer cells to adapt to drastic changes in the tumor environment. Tumors adapt to conventional antineoplastic therapies by generating chemoresistance, residual disease, and tumor relapse [7]. Although its role is not fully understood, it is considered a promising therapeutic target in the fight against cancer.
2. Metabolic Pathways in Cancer
Metabolic reprogramming is a common and essential cancer feature. Contrary to what has been considered in the past, this process is not stable and tends to vary according to the availability of substrates and the different metabolic demands for cell proliferation, growth, invasion, and survival [8]. Cancer cells alter their ability to metabolize carbohydrates, lipids, and proteins to adapt to cellular demands, environmental conditions, and changing accessible substrates [9]. However, substrate unavailability is not the only inducer of this hallmark. It has been shown that the tumor microenvironment plays a significant role in the metabolic reprogramming of cancer cells and tumor environment-dependent adaptation [10].
2.1. Carbohydrate Metabolism
Increased glucose uptake and lactate production in the presence of oxygen, regardless of the anabolic and catabolic needs of cancer cells, has been recognized as a cancer trademark for nearly a century. This is reflected in the coupling of NAD/NADH between glyceraldehyde phosphate dehydrogenase and LDH [11]. On the other hand, mitochondrial uncoupling in cancer cells represents an important source of NAD in the cytosol, increasing glycolytic efflux. An incremented flow of glucose provides intermediaries for different cellular pathways, among which the PPP pathway stands out, since it is the most studied, where G6PD and 6-phosphogluconate dehydrogenase participates in redox homeostasis through the generation of GSH [12] and the synthesis of glycerol-3-phosphate for lipid biosynthesis (Figure 1) [13]. However, other critical metabolic pathways exist, including nuclear glycogen metabolism, which is increased in specific cancer subtypes [14], and gluconeogenesis, which plays an essential role during glucose deprivation through the metabolic flexibility conferred by PCK1 or PCK2, promoting cancer cell survival [15].
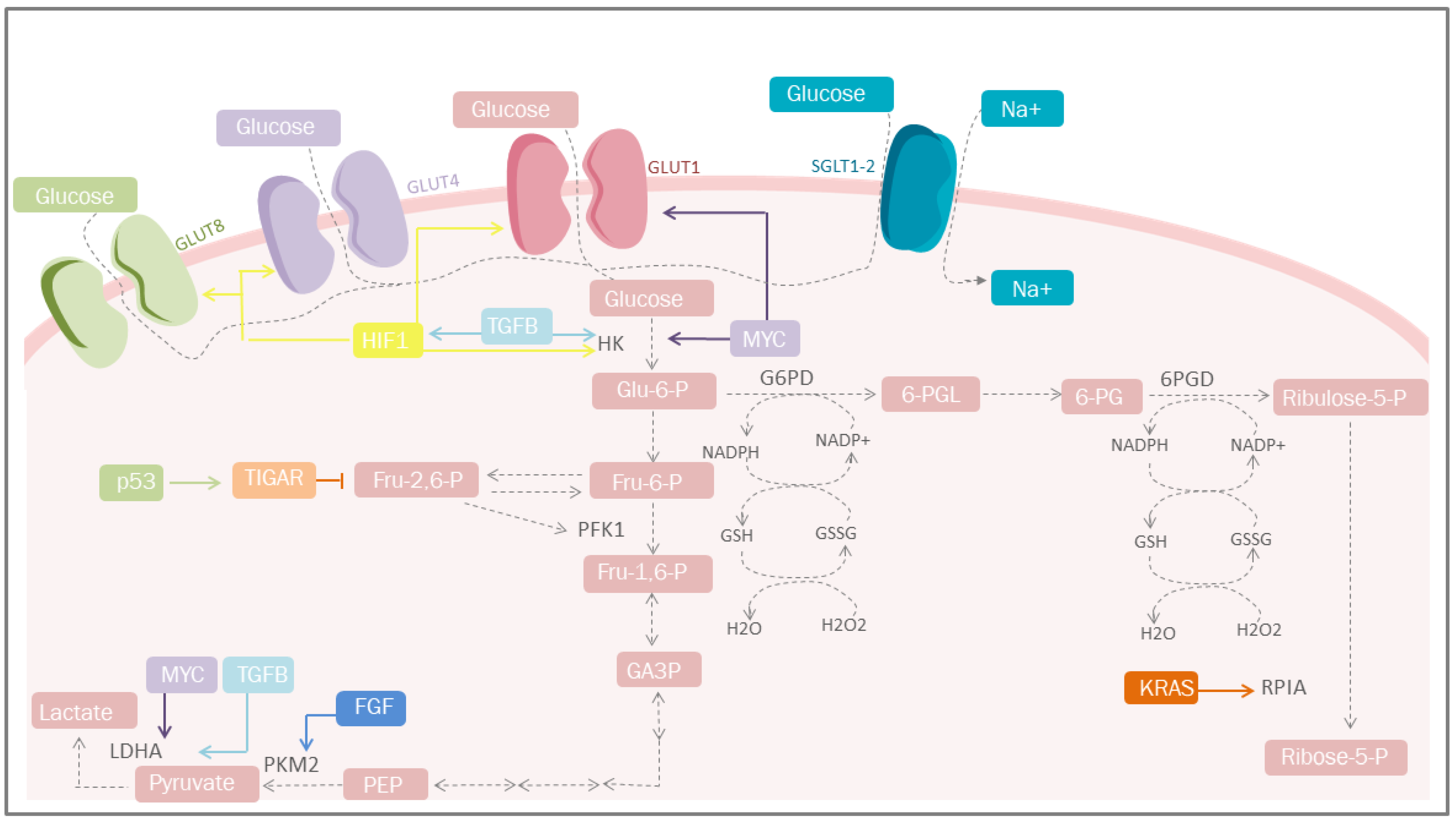 Figure 1. Carbohydrate metabolism and mediators of metabolic reprogramming. Cancer cells must acquire a greater amount of nutrients, especially glucose. One of the mediators is HIF-1, which increases glucose uptake through the induction of GLUT-1, GLUT-4 and GLUT-1; simultaneosly, it can also be stimulated by TGF-β through the PI3K/AKT/mTOR pathway. Other important mediators are p53 that plays a protective role against ROS. GLUT, glucose transporters; Glu-6-P, glucose 6-phosphate; Fru-6-P, fructose 6-bisphosphate; Fru-1,6-P, fructose 1,6-bisphosphate; Fru-2,6-P, fructose 2,6-bisphosphate; GA-3-P, glyceraldehyde 3-phosphate; PEP, phosphoenolpyruvate; HK, hexokinase; TGFB, transforming growth factor beta; PFK1, phosphofructokinase 1; PKM2, pyruvate kinase M2; LDHA, lactate dehydrogenase A; G6PD, glucose-6-phosphate dehydrogenase; 6-PGL, 6-phosphogluconolactonase; NADP+, nicotinamide adenine dinucleotide phosphate; NADPH, reduced form of NADP; GSSG, glutathione disulfide; GSH, glutathione; H2O2, hydrogen peroxide; TIGAR, Tp53-induced glycolysis and apoptosis regulator; FGF, fibroblast growth factor; HIF-1, hypoxia-inducible factor 1; PPP, pentose phosphate pathway; KRAS, Kirsten-ras; RPIA, ribose-5 phosphate isomerase; MYC, proto-oncogene; Ribulose-5-P, ribulose 5-phosphate; Ribose-5-P, ribose 5-phosphate; SGLT1/2, sodium-glucose cotransporter-1/2.
Figure 1. Carbohydrate metabolism and mediators of metabolic reprogramming. Cancer cells must acquire a greater amount of nutrients, especially glucose. One of the mediators is HIF-1, which increases glucose uptake through the induction of GLUT-1, GLUT-4 and GLUT-1; simultaneosly, it can also be stimulated by TGF-β through the PI3K/AKT/mTOR pathway. Other important mediators are p53 that plays a protective role against ROS. GLUT, glucose transporters; Glu-6-P, glucose 6-phosphate; Fru-6-P, fructose 6-bisphosphate; Fru-1,6-P, fructose 1,6-bisphosphate; Fru-2,6-P, fructose 2,6-bisphosphate; GA-3-P, glyceraldehyde 3-phosphate; PEP, phosphoenolpyruvate; HK, hexokinase; TGFB, transforming growth factor beta; PFK1, phosphofructokinase 1; PKM2, pyruvate kinase M2; LDHA, lactate dehydrogenase A; G6PD, glucose-6-phosphate dehydrogenase; 6-PGL, 6-phosphogluconolactonase; NADP+, nicotinamide adenine dinucleotide phosphate; NADPH, reduced form of NADP; GSSG, glutathione disulfide; GSH, glutathione; H2O2, hydrogen peroxide; TIGAR, Tp53-induced glycolysis and apoptosis regulator; FGF, fibroblast growth factor; HIF-1, hypoxia-inducible factor 1; PPP, pentose phosphate pathway; KRAS, Kirsten-ras; RPIA, ribose-5 phosphate isomerase; MYC, proto-oncogene; Ribulose-5-P, ribulose 5-phosphate; Ribose-5-P, ribose 5-phosphate; SGLT1/2, sodium-glucose cotransporter-1/2.In the absence of glucose, cancer cells overexpress phosphoenolpyruvate carboxykinase, initiate gluconeogenesis, and metabolize glucose [16]. Other carbohydrates that play essential roles during cancer stages include hexosamine and fructose. Hexosamine promotes EMT by increasing the nuclear location of beta-catenin by activating its pathway through the glutamine-fructose-6-phosphate transaminase [17]. In contrast, fructose drives metabolic reprogramming by promoting central carbon metabolism via aldolase B to induce metastasis [18].
2.2. Lipid Metabolism
Even though the most studied aspect of metabolic reprogramming concerns glucose, the altered metabolism of lipids has received much attention due to their essential role as structural components of the cellular membrane, second messengers, and a source of fuel for energy production [19].
During cancer, reprogramming of lipid metabolism results from several processes, such as increased fatty acid uptake, de novo lipogenesis, beta-oxidation of fatty acids, and altered lipid storage [20]. In cancer lipid metabolism, a common feature is high levels of fatty acid uptake due to the presence of specialized transporters in the plasma membrane. CD36, fatty acid transport protein family (FATPs), and plasma membrane fatty acid-binding proteins (FABPpm) all exacerbate the aggressiveness, growth, and survival of cancer cells [20] (Figure 2).
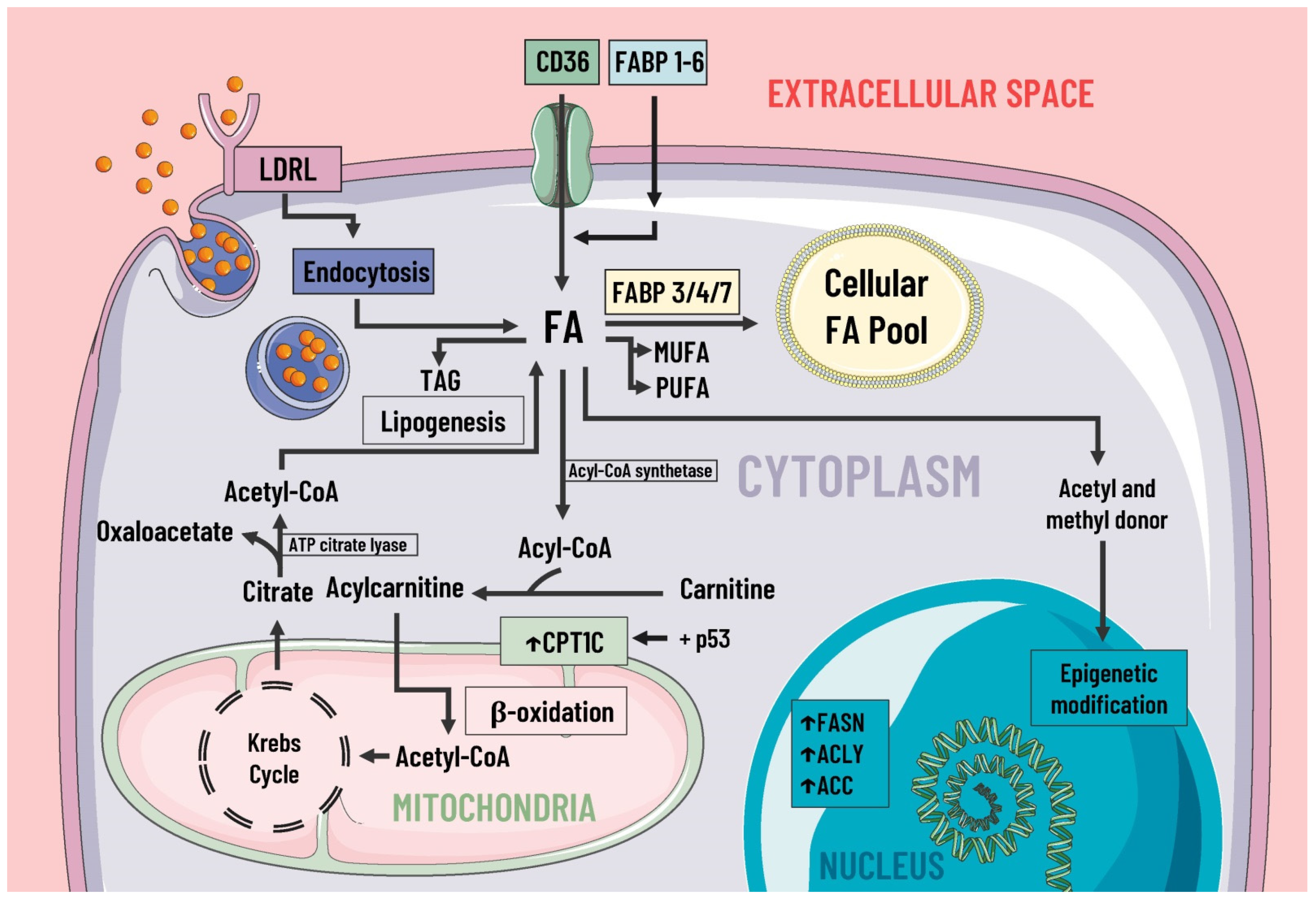 Figure 2. Lipids metabolism and mediators of metabolic reprogramming. Lipid metabolism in cancer cells is altered to increase the availability of these molecules, which provide structural components for the cell membrane, second messengers, and a fuel source. Fatty acids enter the cell by several pathways, including LDRL-mediated endocytosis and a wide variety of membrane transporters, such as CD36 and FABP1-6. Another source of FAS is lipogenesis. Once in the intracellular space, FAS can be stored in the cellular FAS pool by means of FABP 3/4/7, and can also serve as a substrate for the formation of MUFA, PUFA, TAG, etc. Likewise, some researchers suggest FAS are a great energy source through β-oxidation and act as donors of acetyl and methyl groups, participating in epigenetic modifications. In cancer, there is an increase in the expression of enzymes involved in lipogenesis, cholesterol synthesis, and beta-oxidation, such as ACC, FASN, ACLY, and CPT1C, where the latter is due to p53. LDRL, low-density lipoprotein receptor; CD36, a cluster of differentiation 36; FABP, fatty acid-binding protein; FAS, fatty acids synthetase; MUFA, monounsaturated fatty acids; PUFA, polyunsaturated fatty acids; TAG, triacylglycerides; CPT1C, carnitine palmitoyltransferase 1C; FASN, fatty acid synthase; ACLY, ATP citrate lyase; ACC, acetyl-CoA carboxylase; ↑, increase.
Figure 2. Lipids metabolism and mediators of metabolic reprogramming. Lipid metabolism in cancer cells is altered to increase the availability of these molecules, which provide structural components for the cell membrane, second messengers, and a fuel source. Fatty acids enter the cell by several pathways, including LDRL-mediated endocytosis and a wide variety of membrane transporters, such as CD36 and FABP1-6. Another source of FAS is lipogenesis. Once in the intracellular space, FAS can be stored in the cellular FAS pool by means of FABP 3/4/7, and can also serve as a substrate for the formation of MUFA, PUFA, TAG, etc. Likewise, some researchers suggest FAS are a great energy source through β-oxidation and act as donors of acetyl and methyl groups, participating in epigenetic modifications. In cancer, there is an increase in the expression of enzymes involved in lipogenesis, cholesterol synthesis, and beta-oxidation, such as ACC, FASN, ACLY, and CPT1C, where the latter is due to p53. LDRL, low-density lipoprotein receptor; CD36, a cluster of differentiation 36; FABP, fatty acid-binding protein; FAS, fatty acids synthetase; MUFA, monounsaturated fatty acids; PUFA, polyunsaturated fatty acids; TAG, triacylglycerides; CPT1C, carnitine palmitoyltransferase 1C; FASN, fatty acid synthase; ACLY, ATP citrate lyase; ACC, acetyl-CoA carboxylase; ↑, increase.CD36 facilitates the uptake of fatty acids by tumor cells, enhances lipid droplet (LD) development, and promotes the formation of acylcarnitine, monoacylglycerols, and other phospholipids [21]. It also facilitates fatty acid oxidation (FAO) [21]. The increase in fatty acid uptake hampers antigen presentation. Considering its high heterogeneity and binding capacity for different ligands, CD36 is intimately related to the presence of thrombospondin-1 (TSP-1), known for its antitumorigenic effects. Current evidence has shown that it also plays an essential part in cancer development by promoting the formation of an embolus of tumor cells and cell adhesion. In addition, it induces the release of TGF-β, promoting tumor growth by altering angiogenesis through the modulation of proangiogenic proteins, such as JNK and p38a [21][22].
In cancer cells, lipogenesis parallels elevated FAS expression, cell migration, evasion of apoptosis, and aggressiveness [23][24]. The accumulation of fatty acids by lipogenesis results in their storage in LDs, and cytoplasmic lipases catalyze their output. Adipose triglyceride lipase (ATGL), hormone-sensitive lipase (HSL), and monoacylglycerol lipase (MAGL) sustain tumor survival [25].
The activation of lipases and the availability of fatty acids increases the rate of electron flow. It produces an increase in the formation of ROS, which have multiple protumorigenic effects by facilitating cell proliferation, tumor growth, and adaptation through their participation in multiple signaling pathways, among which stand out the following: the mitogen-activated protein kinase (MAPK) cascades, phosphoinositide-3-kinase (PI3K)/Akt, and the nuclear factor kappa light chain-enhancer of activated B cell (NF-kB) pathways. Therefore, the correlation between lipid catabolism and ROS production is important in regulating cancer growth and progression [25][26].
All the effects mentioned above are augmented by the loss of p53 activity and mutations [27]. The upregulation of enzymes related to mevalonate anabolism are involved in cholesterol synthesis and protein prenylation [28].
2.3. Protein Metabolism
Cancer cells adapt their metabolism to support biomass production; therefore, they have a high rate of protein synthesis. To maintain correct protein synthesis, the contribution of amino acids is vital as proteogenic building blocks. Among the most notable is glutamine, the most abundant non-essential amino acid in plasma and the main nitrogen donor for the synthesis of purines, pyrimidines, and NAD [29], so its deficiency leads to the interruption of protein synthesis [30] (Figure 3).
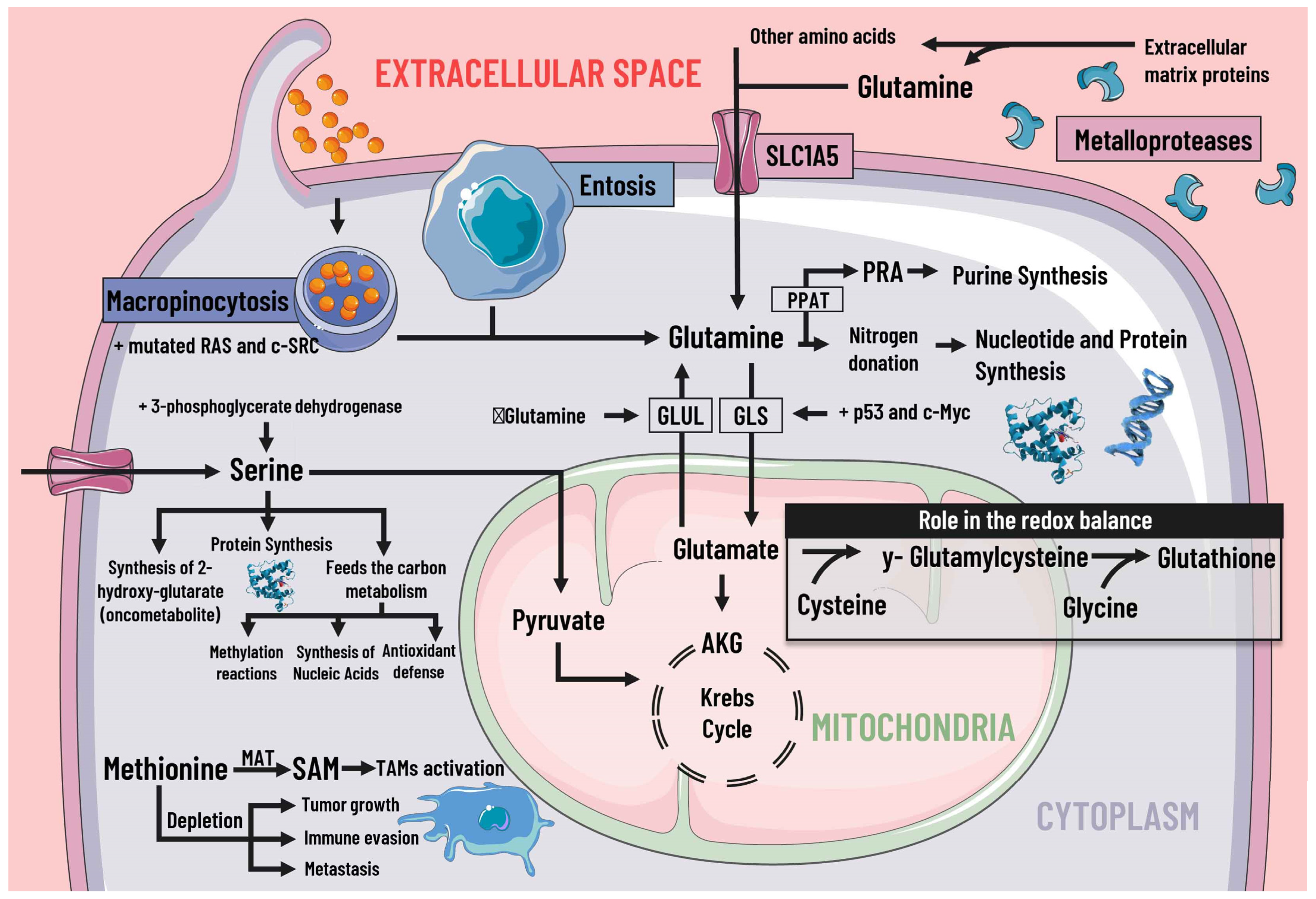 Figure 3. Protein metabolism and mediators of metabolic reprogramming. Protein metabolism in cancer cells is altered due to the immense demand generated by constant cell division, which is why amino acids are of great importance as proteogenic building blocks. Among the amino acids, glutamine is of great relevance in cancer cells. Glutamine enters the cell by different pathways, including macropinocytosis (which is increased by mutated RAS and c-SRC), entosis, degradation of cell-matrix proteins by metalloproteases, and transport by SLC1A5. Glutamine serves as a nitrogen donor and is used to synthesise nucleotides, purines, and proteins. It can also be a source of energy through glutaminolysis, which p53 and c-Myc stimulate. When there is a decrease in glutamine levels, it is synthesized by GLUL using glutamate as a substrate. Likewise, glutamine intervenes indirectly in the redox balance through glutamate. Serine is also essential, since increases in serine and 3-phosphoglycerate dehydrogenase, the first enzyme involved in its synthesis, are associated with tumor growth. Serine feeds carbon metabolism and protein synthesis, favors the synthesis of the oncometabolite 2-hydroxy-glutarate, and can act as an energy source, since it is an anaplerotic metabolite. In addition, methionine and SAM facilitate the pattern of histone methylation in monocytes/macrophages and the activation of TAMs. Depletion of exogenous methionine promotes tumor growth, metastasis, and immune evasion. SLC1A5 solute transporter family member 5; PPAT, phosphoribosyl pyrophosphate amidotransferase; PRA, phosphoribosylamine; GLS, glutaminase; GLUL, glutamine synthetase; AKG, alpha-ketoglutarate; SAMs S-adenosylmethionine; MAT, methionine adenosyltransferase; TAMs, tumor-associated macrophages.
Figure 3. Protein metabolism and mediators of metabolic reprogramming. Protein metabolism in cancer cells is altered due to the immense demand generated by constant cell division, which is why amino acids are of great importance as proteogenic building blocks. Among the amino acids, glutamine is of great relevance in cancer cells. Glutamine enters the cell by different pathways, including macropinocytosis (which is increased by mutated RAS and c-SRC), entosis, degradation of cell-matrix proteins by metalloproteases, and transport by SLC1A5. Glutamine serves as a nitrogen donor and is used to synthesise nucleotides, purines, and proteins. It can also be a source of energy through glutaminolysis, which p53 and c-Myc stimulate. When there is a decrease in glutamine levels, it is synthesized by GLUL using glutamate as a substrate. Likewise, glutamine intervenes indirectly in the redox balance through glutamate. Serine is also essential, since increases in serine and 3-phosphoglycerate dehydrogenase, the first enzyme involved in its synthesis, are associated with tumor growth. Serine feeds carbon metabolism and protein synthesis, favors the synthesis of the oncometabolite 2-hydroxy-glutarate, and can act as an energy source, since it is an anaplerotic metabolite. In addition, methionine and SAM facilitate the pattern of histone methylation in monocytes/macrophages and the activation of TAMs. Depletion of exogenous methionine promotes tumor growth, metastasis, and immune evasion. SLC1A5 solute transporter family member 5; PPAT, phosphoribosyl pyrophosphate amidotransferase; PRA, phosphoribosylamine; GLS, glutaminase; GLUL, glutamine synthetase; AKG, alpha-ketoglutarate; SAMs S-adenosylmethionine; MAT, methionine adenosyltransferase; TAMs, tumor-associated macrophages.The high rate of glutamine consumption and uptake through the SLC1A5 transporter shows that cancer cells are dependent on this amino acid. Glutamine provides intermediates for the TCA cycle. In addition, it is also involved in protein synthesis by being the precursor of other amino acids, such as glutamate, asparagine, aspartate, and proline [31]. Some mediators can regulate glutamine metabolism, such as p53 and c-Myc, by stimulating GLS [32]. The deamination of glutamine leads to the release of ammonia. Ammonia is recycled for generating other amino acids, including aspartate, leucine, and proline, through GLUD [33]. When extracellular glutamine is scarce, cancer cells can increase their production through the enzyme glutamate-ammonia ligase (GLUL) [29].
Another amino acid of great importance in cancer development is arginine, an essential amino acid that participates in many metabolic pathways. In cancer cells, the enzyme argininosuccinate synthetase (ASS) (theh limiting enzyme for de novo arginine synthesis) is deficient, which leads the tumor cell to obtain exogenous arginine [34]. On the other hand, serine contributes to the donation of metabolites destined for the TCA cycle and favors the synthesis of oncometabolites [35]. Likewise, other essential amino acids, including aspartate and proline, support nucleotide biosynthesis and ATP generation [30].
Cancer cells use alternative processes for extracellular matrix (ECM) survival and proliferation [9]. These processes include the degradation of ECM proteins into peptides by matrix metalloproteases. Macropinocytosis is also used to internalize and degrade exogenous proteins and entosis. One living cell is completely internalized by another, where it is destroyed and digested by the host cell [36].
2.4. Methionine, S-Adenosylmethionine and Cysteine
Similarly, methionine and S-adenosylmethionine (SAM), a biological methyl donor generated from methionine by methionine adenosyltransferase (MAT) enzymes, facilitate histone methylation patterns in monocytes/macrophages as well as activation of tumor-associated macrophages (TAMs). It is known that depletion of exogeneous methionine or inhibition of MAT2A impairs M1 and M2 polarization of macrophages in addition to promoting tumor growth, metastasis, and immune evasion [37].
At the same time, the relationship between cysteine and cancer has been actively studied. It is acknowledged that sulfur and carbon metabolism reprogramming underlies the adaption to a hypoxic microenvironment promoted by cysteine. In the extracellular environment, cysteine is present in an oxidized dipeptide named cystine. Cystine uptake is driven by the cystine/glutamate antiporter xCT and immediately reduced to cysteine by NADPH. Furthermore, cysteine is synthesized de novo by the transsulfuration (TSS) pathway in cancer cells under cystine deprivation [38].
2.5. Isoenzymes
2.5.1. Fumarate Hydratase (FH)
The enzyme FH is part of the TCA cycle, where it catalyzes the reversible formation of fumarate to malate. Its mutation or silencing contributes to carcinogenesis by compensating for mitochondrial deterioration by metabolic changes [39]. FH increases glycolytic flux. Instead of oxidizing glucose in the mitochondria, it is diverted to the PPP pathway, and the rest is converted into lactate through increased glycolytic enzymes and inhibition of PDH [40]. In addition, due to the decreased entry of glucose into the TCA cycle, it is replaced by glutamine as the primary carbon source to supply alpha-ketoglutarate and subsequently generate NADPH destined to participate in oxidative phosphorylation, along with the transformation to citrate for lipid biosynthesis thanks to reductive carboxylation [41].
The accumulation of intracellular fumarate can create a state of pseudohypoxia through HIF stabilization via the NF-kB pathway. The intracellular increase of this oncometabolite pressures cancer cells to metabolize arginine to form argininosuccinate by the enzyme argininosuccinate lyase (ASL) [41].
2.5.2. Isocitrate Dehydrogenase
The enzyme isocitrate dehydrogenase (IDH) is responsible for catalyzing the oxidative decarboxylation of isocitrate to alpha-ketoglutarate and carbon dioxide (CO2) in the TCA pathway. The enzyme is involved in a series of metabolic functions, which includes glucose sensing, glutamine metabolism, lipogenesis, and regulation of the redox state [42].
An IDH mutation leads to major metabolic changes involving increased oxidative stress through the reduction of NADPH [43], lactate dehydrogenase A (LDHA) silencing (suggesting that the carbon source is preferentially derived from alpha-ketoglutarate via glutamine metabolism), and PDH suppression (via elevated PDK3 expression) accompanied by increased activity of PC, an essential enzyme for producing intermediate metabolites in various pathways crucial for gluconeogenesis and lipogenesis [44]. Likewise, an IDH mutation increases 2-hydroxyglutarate, a key metabolite during metabolic reprogramming [45].
2.5.3. Glutaminase
Glutamine is one of the main energy sources used by cancer cells, right after glucose. Glutamine plays a part in metabolic pathways essential for growth, proliferation, and defence against oxidative stress [46]. Its metabolic products, glutamate and alpha-ketoglutarate, participate in synthesising fatty acids, transamination, amino acid synthesis, and TCA support. All the processes mentioned above are metabolized by GLS [47].
There are two GLS isoenzymes: GLS and GLS2. GLS, regulated by the MYC and mTORC-1 oncogenes, engages in macromolecule synthesis as well as energy supply through TCA glutaminolysis, whereas GLS2, regulated by p53, fulfils different roles in energy metabolism and antioxidant defence [48]. Since GLS catabolizes glutamine for the final generation of ATP and GSH, its reduction or silencing play a key role cell proliferation and survival, leading to cell death due to decreased ATP and excessive generation of ROS [49].
2.5.4. Lactate Dehydrogenases
In cancer, most of the pyruvate generated by excessive glycolysis is diverted to the generation of lactate, even if oxygen is present, with the help of LDH. One way in which LDH is able to facilitate tumor progression is through interaction with the tumor stroma, where it is important for ECM remodeling, activation of signaling pathways, the release of inflammatory molecules, and fueling of CAFs, all thanks to the lactate generated in the tumor cell that subsequently released by increased expression of the lactate transporter MCT4. An increase in extracellular lactate also triggers an increase in the expression of MCT1 and LDH in the CAFs, which are then absorbed and converted into pyruvate to satisfy the needs of these stromal cells, thereby initiating positive feedback between cancer cells and the stroma [50].
References
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.K.; Ouellette, B.F.F.; Li, C.H.; et al. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93.
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer Statistics for the Year 2020: An Overview. Int. J. Cancer 2021, 149, 778–789.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674.
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46.
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200.
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218.
- da Silva-Diz, V.; Lorenzo-Sanz, L.; Bernat-Peguera, A.; Lopez-Cerda, M.; Muñoz, P. Cancer Cell Plasticity: Impact on Tumor Progression and Therapy Response. Semin. Cancer Biol. 2018, 53, 48–58.
- Lue, H.; Podolak, J.; Kolahi, K.; Cheng, L.; Rao, S.; Garg, D.; Xue, C.-H.; Rantala, J.K.; Tyner, J.W.; Thornburg, K.L.; et al. Metabolic Reprogramming Ensures Cancer Cell Survival despite Oncogenic Signaling Blockade. Genes Dev. 2017, 31, 2067–2084.
- Sun, L.; Suo, C.; Li, S.-T.; Zhang, H.; Gao, P. Metabolic Reprogramming for Cancer Cells and Their Microenvironment: Beyond the Warburg Effect. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 51–66.
- Xia, L.; Oyang, L.; Lin, J.; Tan, S.; Han, Y.; Wu, N.; Yi, P.; Tang, L.; Pan, Q.; Rao, S.; et al. The Cancer Metabolic Reprogramming and Immune Response. Mol. Cancer 2021, 20, 28.
- Li, T.; Tan, X.; Yang, R.; Miao, Y.; Zhang, M.; Xi, Y.; Guo, R.; Zheng, M.; Li, B. Discovery of Novel Glyceraldehyde-3-Phosphate Dehydrogenase Inhibitor via Docking-Based Virtual Screening. Bioorg. Chem. 2020, 96, 103620.
- Ghanbari Movahed, Z.; Rastegari-Pouyani, M.; Mohammadi, M.H.; Mansouri, K. Cancer Cells Change Their Glucose Metabolism to Overcome Increased ROS: One Step from Cancer Cell to Cancer Stem Cell? Biomed. Pharmacother. 2019, 112, 108690.
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47.
- Sun, R.C.; Dukhande, V.V.; Zhou, Z.; Young, L.E.A.; Emanuelle, S.; Brainson, C.F.; Gentry, M.S. Nuclear Glycogenolysis Modulates Histone Acetylation in Human Non-Small Cell Lung Cancers. Cell Metab. 2019, 30, 903–916.
- Grasmann, G.; Smolle, E.; Olschewski, H.; Leithner, K. Gluconeogenesis in Cancer Cells—Repurposing of a Starvation-Induced Metabolic Pathway? Biochim. Biophys. Acta BBA Rev. Cancer 2019, 1872, 24–36.
- Vincent, E.E.; Sergushichev, A.; Griss, T.; Gingras, M.-C.; Samborska, B.; Ntimbane, T.; Coelho, P.P.; Blagih, J.; Raissi, T.C.; Choinière, L.; et al. Mitochondrial Phosphoenolpyruvate Carboxykinase Regulates Metabolic Adaptation and Enables Glucose-Independent Tumor Growth. Mol. Cell 2015, 60, 195–207.
- Zhou, L.; Luo, M.; Cheng, L.-J.; Li, R.-N.; Liu, B.; Linghu, H. Glutamine-Fructose-6-Phosphate Transaminase 2 (GFPT2) Promotes the EMT of Serous Ovarian Cancer by Activating the Hexosamine Biosynthetic Pathway to Increase the Nuclear Location of β-Catenin. Pathol. Res. Pract. 2019, 215, 152681.
- Bu, P.; Chen, K.Y.; Xiang, K.; Johnson, C.; Crown, S.B.; Rakhilin, N.; Ai, Y.; Wang, L.; Xi, R.; Astapova, I.; et al. Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell Metab. 2018, 27, 1249–1262.
- Hammond, G.R.V.; Burke, J.E. Novel Roles of Phosphoinositides in Signaling, Lipid Transport, and Disease. Curr. Opin. Cell Biol. 2020, 63, 57–67.
- Fernández, L.P.; Gómez de Cedrón, M.; Ramírez de Molina, A. Alterations of Lipid Metabolism in Cancer: Implications in Prognosis and Treatment. Front. Oncol. 2020, 10, 577420.
- Wang, J.; Li, Y. CD36 Tango in Cancer: Signaling Pathways and Functions. Theranostics 2019, 9, 4893–4908.
- Tanase, C.; Enciu, A.M.; Codrici, E.; Popescu, I.D.; Dudau, M.; Dobri, A.M.; Pop, S.; Mihai, S.; Gheorghișan-Gălățeanu, A.-A.; Hinescu, M.E. Fatty Acids, CD36, Thrombospondin-1, and CD47 in Glioblastoma: Together and/or Separately? Int. J. Mol. Sci. 2022, 23, 604.
- Hu, J.; Zhang, L.; Chen, W.; Shen, L.; Jiang, J.; Sun, S.; Chen, Z. Role of Intra- and Extracellular Lipid Signals in Cancer Stemness and Potential Therapeutic Strategy. Front. Pharmacol. 2021, 12, 730751.
- Fhu, C.W.; Ali, A. Fatty Acid Synthase: An Emerging Target in Cancer. Molecules 2020, 25, 3935.
- Castelli, S.; De Falco, P.; Ciccarone, F.; Desideri, E.; Ciriolo, M.R. Lipid Catabolism and ROS in Cancer: A Bidirectional Liaison. Cancers 2021, 13, 5484.
- Bartolacci, C.; Andreani, C.; El-Gammal, Y.; Scaglioni, P.P. Lipid Metabolism Regulates Oxidative Stress and Ferroptosis in RAS-Driven Cancers: A Perspective on Cancer Progression and Therapy. Front. Mol. Biosci. 2021, 8, 706650.
- Casals, N.; Zammit, V.; Herrero, L.; Fadó, R.; Rodríguez-Rodríguez, R.; Serra, D. Carnitine Palmitoyltransferase 1C: From Cognition to Cancer. Prog. Lipid Res. 2016, 61, 134–148.
- Parrales, A.; Iwakuma, T. P53 as a Regulator of Lipid Metabolism in Cancer. Int. J. Mol. Sci. 2016, 17, 2074.
- Bott, A.J.; Shen, J.; Tonelli, C.; Zhan, L.; Sivaram, N.; Jiang, Y.-P.; Yu, X.; Bhatt, V.; Chiles, E.; Zhong, H.; et al. Glutamine Anabolism Plays a Critical Role in Pancreatic Cancer by Coupling Carbon and Nitrogen Metabolism. Cell Rep. 2019, 29, 1287–1298.
- Vettore, L.; Westbrook, R.L.; Tennant, D.A. New Aspects of Amino Acid Metabolism in Cancer. Br. J. Cancer 2020, 122, 150–156.
- Hosios, A.M.; Hecht, V.C.; Danai, L.V.; Johnson, M.O.; Rathmell, J.C.; Steinhauser, M.L.; Manalis, S.R.; Vander Heiden, M.G. Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev. Cell 2016, 36, 540–549.
- Kurbegovic, A.; Trudel, M. The Master Regulators Myc and P53 Cellular Signaling and Functions in Polycystic Kidney Disease. Cell. Signal. 2020, 71, 109594.
- Spinelli, J.B.; Yoon, H.; Ringel, A.E.; Jeanfavre, S.; Clish, C.B.; Haigis, M.C. Metabolic Recycling of Ammonia via Glutamate Dehydrogenase Supports Breast Cancer Biomass. Science 2017, 358, 941–946.
- Lukey, M.J.; Katt, W.P.; Cerione, R.A. Targeting Amino Acid Metabolism for Cancer Therapy. Drug Discov. Today 2017, 22, 796–804.
- Corchado-Cobos, R.; García-Sancha, N.; Mendiburu-Eliçabe, M.; Gómez-Vecino, A.; Jiménez-Navas, A.; Pérez-Baena, M.J.; Holgado-Madruga, M.; Mao, J.-H.; Cañueto, J.; Castillo-Lluva, S.; et al. Pathophysiological Integration of Metabolic Reprogramming in Breast Cancer. Cancers 2022, 14, 322.
- Durgan, J.; Florey, O. Cancer Cell Cannibalism: Multiple Triggers Emerge for Entosis. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 831–841.
- Zhang, Y.; Yang, H.; Zhao, J.; Wan, P.; Hu, Y.; Lv, K.; Hu, Y.; Yang, X.; Ma, M. Activation of MAT2A-RIP1 Signaling Axis Reprograms Monocytes in Gastric Cancer. J. Immunother. Cancer 2021, 9, e001364.
- Zhang, H.-F.; Klein Geltink, R.I.; Parker, S.J.; Sorensen, P.H. Transsulfuration, Minor Player or Crucial for Cysteine Homeostasis in Cancer. Trends Cell Biol. 2022, S0962-8924(22)000605.
- Tyrakis, P.A.; Yurkovich, M.E.; Sciacovelli, M.; Papachristou, E.K.; Bridges, H.R.; Gaude, E.; Schreiner, A.; D’Santos, C.; Hirst, J.; Hernandez-Fernaud, J.; et al. Fumarate Hydratase Loss Causes Combined Respiratory Chain Defects. Cell Rep. 2017, 21, 1036–1047.
- Gonçalves, E.; Sciacovelli, M.; Costa, A.S.H.; Tran, M.G.B.; Johnson, T.I.; Machado, D.; Frezza, C.; Saez-Rodriguez, J. Post-Translational Regulation of Metabolism in Fumarate Hydratase Deficient Cancer Cells. Metab. Eng. 2018, 45, 149–157.
- Schmidt, S.; Gay, D.; Uthe, F.W.; Denk, S.; Paauwe, M.; Matthes, N.; Diefenbacher, M.E.; Bryson, S.; Warrander, F.C.; Erhard, F.; et al. A MYC–GCN2–EIF2α Negative Feedback Loop Limits Protein Synthesis to Prevent MYC-Dependent Apoptosis in Colorectal Cancer. Nat. Cell Biol. 2019, 21, 1413–1424.
- Masui, K.; Onizuka, H.; Cavenee, W.K.; Mischel, P.S.; Shibata, N. Metabolic Reprogramming in the Pathogenesis of Glioma: Update. Neuropathol. Off. J. Jpn. Soc. Neuropathol. 2019, 39, 3–13.
- Gelman, S.J.; Naser, F.; Mahieu, N.G.; McKenzie, L.D.; Dunn, G.P.; Chheda, M.G.; Patti, G.J. Consumption of NADPH for 2-HG Synthesis Increases Pentose Phosphate Pathway Flux and Sensitizes Cells to Oxidative Stress. Cell Rep. 2018, 22, 512–522.
- Izquierdo-Garcia, J.L.; Viswanath, P.; Eriksson, P.; Cai, L.; Radoul, M.; Chaumeil, M.M.; Blough, M.; Luchman, H.A.; Weiss, S.; Cairncross, J.G.; et al. IDH1 Mutation Induces Reprogramming of Pyruvate Metabolism. Cancer Res. 2015, 75, 2999–3009.
- Fitzpatrick, S.F.; Lambden, S.; Macias, D.; Puthucheary, Z.; Pietsch, S.; Mendil, L.; McPhail, M.J.W.; Johnson, R.S. 2-Hydroxyglutarate Metabolism Is Altered in an In Vivo Model of LPS Induced Endotoxemia. Front. Physiol. 2020, 11, 1–8.
- Jiang, J.; Srivastava, S.; Zhang, J. Starve Cancer Cells of Glutamine: Break the Spell or Make a Hungry Monster? Cancers 2019, 11, 804.
- Masisi, B.K.; El Ansari, R.; Alfarsi, L.; Rakha, E.A.; Green, A.R.; Craze, M.L. The Role of Glutaminase in Cancer. Histopathology 2020, 76, 498–508.
- Katt, W.P.; Lukey, M.J.; Cerione, R.A. A Tale of Two Glutaminases: Homologous Enzymes with Distinct Roles in Tumorigenesis. Future Med. Chem. 2017, 9, 223–243.
- Matés, J.M.; Campos-Sandoval, J.A.; Márquez, J. Glutaminase Isoenzymes in the Metabolic Therapy of Cancer. Biochim. Biophys. Acta BBA Rev. Cancer 2018, 1870, 158–164.
- Gouirand, V.; Guillaumond, F.; Vasseur, S. Influence of the Tumor Microenvironment on Cancer Cells Metabolic Reprogramming. Front. Oncol. 2018, 8, 117.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.8K
Revisions:
2 times
(View History)
Update Date:
27 Jun 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No