Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Angel Ortega and Version 2 by Camila Xu.
Reprogramming is a process mediated by multiple factors, including oncogenes, growth factors, hypoxia-induced factors, and the loss of suppressor gene function, which support malignant transformation and tumor development in addition to cell heterogeneity. Consequently, this hallmark promotes resistance to conventional anti-tumor therapies by adapting to the drastic changes in the nutrient microenvironment that these therapies entail. Therefore, it represents a revolutionary landscape during cancer progression that could be useful for developing new and improved therapeutic strategies targeting alterations in cancer cell metabolism, such as the deregulated mTOR and PI3K pathways.
- metabolic reprogramming
- tumor microenvironment
- energy metabolism
1. Introduction
Cancer is a term used to define a group of diseases characterized by abnormal cell growth with an autonomous and uncontrolled expansion [1]. It represents a significant and escalating public health issue, responsible for one in six deaths worldwide. In this regard, in 2020, 19.3 million new cases and 10.0 million deaths from cancer were estimated [2], numbers that continue to increase, such that by the year 2040, cancer could be diagnosed in more than 29.4 million people per year [3].
It is currently known that cancer cells develop numerous properties that differentiate them from non-cancerous cells. These properties are known as hallmarks and include active proliferation, evasion of growth suppressors, resistance to cell death, angiogenesis, invasion and metastasis, immune evasion, tumour-promoting inflammation, genomic instability, mutation, non-mutational epigenetic reprogramming, polymorphic microbiomes, phenotypic plasticity, and metabolic reprogramming [3][4][3,4]. The latter refers to the ability of cancer cells to modify their metabolism, increasing the absorption and use of carbohydrates, lipids, and proteins to provide a pro-tumorigenic response in the face of the acquisition and maintenance of malignant properties [5].
Many factors mediate metabolic reprogramming: oncogenes, growth factors, hypoxia-induced factors, and the dysfunction of tumor suppressor genes. These alterations cause alterations in cell metabolism, especially glucose, whose absorption rate drastically increases in cancer [6].
Metabolic reprogramming allows cancer cells to adapt to drastic changes in the tumor environment. Tumors adapt to conventional antineoplastic therapies by generating chemoresistance, residual disease, and tumor relapse [7]. Although its role is not fully understood, it is considered a promising therapeutic target in the fight against cancer.
2. Metabolic Pathways in Cancer
Metabolic reprogramming is a common and essential cancer feature. Contrary to what has been considered in the past, this process is not stable and tends to vary according to the availability of substrates and the different metabolic demands for cell proliferation, growth, invasion, and survival [8][19]. Cancer cells alter their ability to metabolize carbohydrates, lipids, and proteins to adapt to cellular demands, environmental conditions, and changing accessible substrates [9][20]. However, substrate unavailability is not the only inducer of this hallmark. It has been shown that the tumor microenvironment plays a significant role in the metabolic reprogramming of cancer cells and tumor environment-dependent adaptation [10][21].2.1. Carbohydrate Metabolism
Increased glucose uptake and lactate production in the presence of oxygen, regardless of the anabolic and catabolic needs of cancer cells, has been recognized as a cancer trademark for nearly a century. This is reflected in the coupling of NAD/NADH between glyceraldehyde phosphate dehydrogenase and LDH [11][22]. On the other hand, mitochondrial uncoupling in cancer cells represents an important source of NAD in the cytosol, increasing glycolytic efflux. An incremented flow of glucose provides intermediaries for different cellular pathways, among which the PPP pathway stands out, since it is the most studied, where G6PD and 6-phosphogluconate dehydrogenase participates in redox homeostasis through the generation of GSH [12][23] and the synthesis of glycerol-3-phosphate for lipid biosynthesis (Figure 1) [13][24]. However, other critical metabolic pathways exist, including nuclear glycogen metabolism, which is increased in specific cancer subtypes [14][25], and gluconeogenesis, which plays an essential role during glucose deprivation through the metabolic flexibility conferred by PCK1 or PCK2, promoting cancer cell survival [15][26].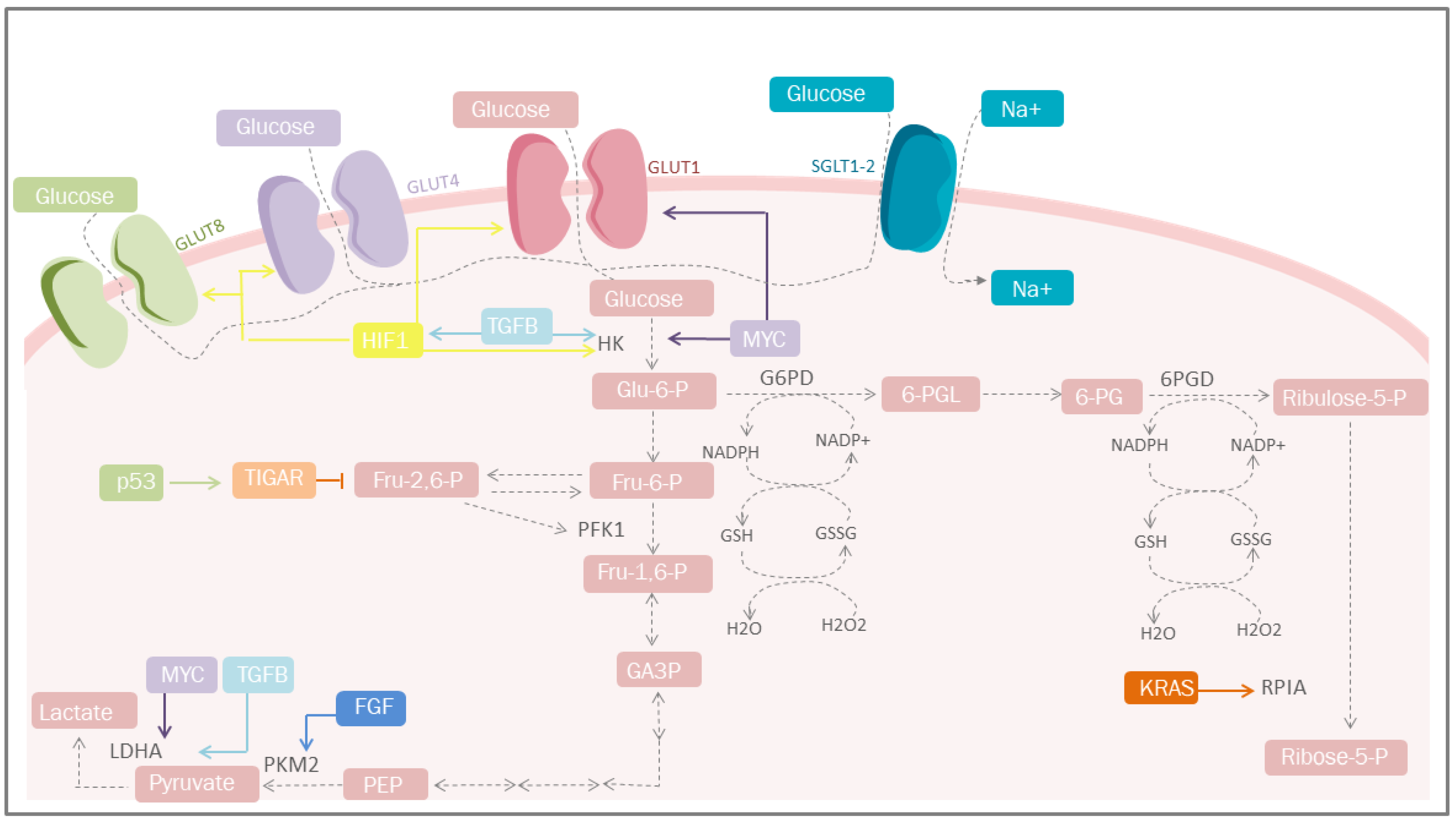
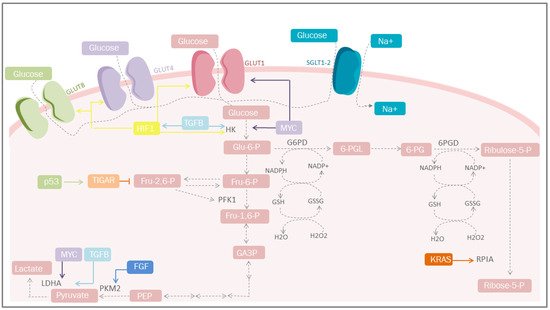
Figure 1. Carbohydrate metabolism and mediators of metabolic reprogramming. Cancer cells must acquire a greater amount of nutrients, especially glucose. One of the mediators is HIF-1, which increases glucose uptake through the induction of GLUT-1, GLUT-4 and GLUT-1; simultaneosly, it can also be stimulated by TGF-β through the PI3K/AKT/mTOR pathway. Other important mediators are p53 that plays a protective role against ROS. GLUT, glucose transporters; Glu-6-P, glucose 6-phosphate; Fru-6-P, fructose 6-bisphosphate; Fru-1,6-P, fructose 1,6-bisphosphate; Fru-2,6-P, fructose 2,6-bisphosphate; GA-3-P, glyceraldehyde 3-phosphate; PEP, phosphoenolpyruvate; HK, hexokinase; TGFB, transforming growth factor beta; PFK1, phosphofructokinase 1; PKM2, pyruvate kinase M2; LDHA, lactate dehydrogenase A; G6PD, glucose-6-phosphate dehydrogenase; 6-PGL, 6-phosphogluconolactonase; NADP+, nicotinamide adenine dinucleotide phosphate; NADPH, reduced form of NADP; GSSG, glutathione disulfide; GSH, glutathione; H2O2, hydrogen peroxide; TIGAR, Tp53-induced glycolysis and apoptosis regulator; FGF, fibroblast growth factor; HIF-1, hypoxia-inducible factor 1; PPP, pentose phosphate pathway; KRAS, Kirsten-ras; RPIA, ribose-5 phosphate isomerase; MYC, proto-oncogene; Ribulose-5-P, ribulose 5-phosphate; Ribose-5-P, ribose 5-phosphate; SGLT1/2, sodium-glucose cotransporter-1/2.
2.2. Lipid Metabolism
Even though the most studied aspect of metabolic reprogramming concerns glucose, the altered metabolism of lipids has received much attention due to their essential role as structural components of the cellular membrane, second messengers, and a source of fuel for energy production [19][30]. During cancer, reprogramming of lipid metabolism results from several processes, such as increased fatty acid uptake, de novo lipogenesis, beta-oxidation of fatty acids, and altered lipid storage [20][17]. In cancer lipid metabolism, a common feature is high levels of fatty acid uptake due to the presence of specialized transporters in the plasma membrane. CD36, fatty acid transport protein family (FATPs), and plasma membrane fatty acid-binding proteins (FABPpm) all exacerbate the aggressiveness, growth, and survival of cancer cells [20][17] (Figure 2).
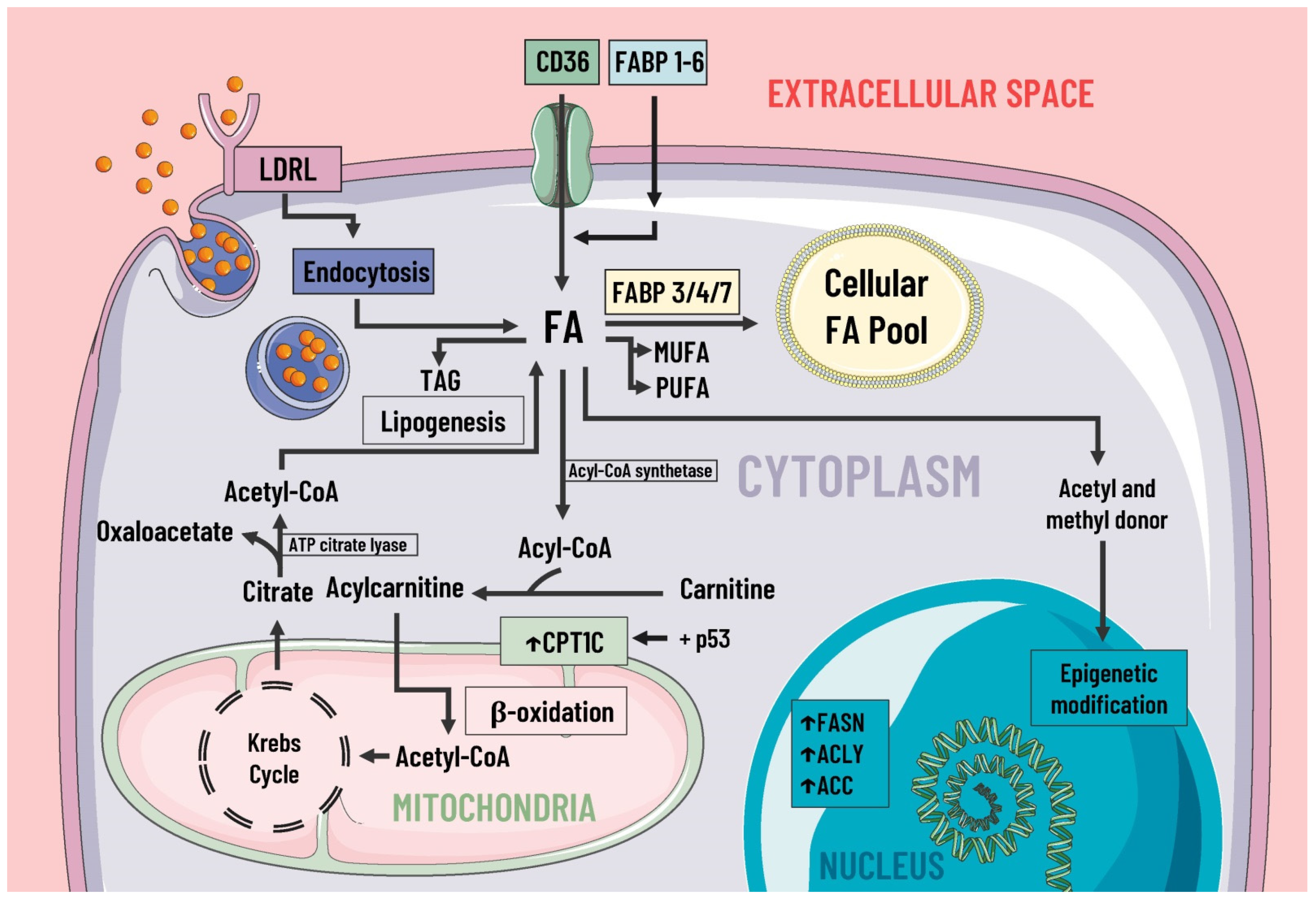
Figure 2. Lipids metabolism and mediators of metabolic reprogramming. Lipid metabolism in cancer cells is altered to increase the availability of these molecules, which provide structural components for the cell membrane, second messengers, and a fuel source. Fatty acids enter the cell by several pathways, including LDRL-mediated endocytosis and a wide variety of membrane transporters, such as CD36 and FABP1-6. Another source of FAS is lipogenesis. Once in the intracellular space, FAS can be stored in the cellular FAS pool by means of FABP 3/4/7, and can also serve as a substrate for the formation of MUFA, PUFA, TAG, etc. Likewise, some researcheuthors suggest FAS are a great energy source through β-oxidation and act as donors of acetyl and methyl groups, participating in epigenetic modifications. In cancer, there is an increase in the expression of enzymes involved in lipogenesis, cholesterol synthesis, and beta-oxidation, such as ACC, FASN, ACLY, and CPT1C, where the latter is due to p53. LDRL, low-density lipoprotein receptor; CD36, a cluster of differentiation 36; FABP, fatty acid-binding protein; FAS, fatty acids synthetase; MUFA, monounsaturated fatty acids; PUFA, polyunsaturated fatty acids; TAG, triacylglycerides; CPT1C, carnitine palmitoyltransferase 1C; FASN, fatty acid synthase; ACLY, ATP citrate lyase; ACC, acetyl-CoA carboxylase; ↑, increase.
2.3. Protein Metabolism
Cancer cells adapt their metabolism to support biomass production; therefore, they have a high rate of protein synthesis. To maintain correct protein synthesis, the contribution of amino acids is vital as proteogenic building blocks. Among the most notable is glutamine, the most abundant non-essential amino acid in plasma and the main nitrogen donor for the synthesis of purines, pyrimidines, and NAD [29][38], so its deficiency leads to the interruption of protein synthesis [30][39] (Figure 3).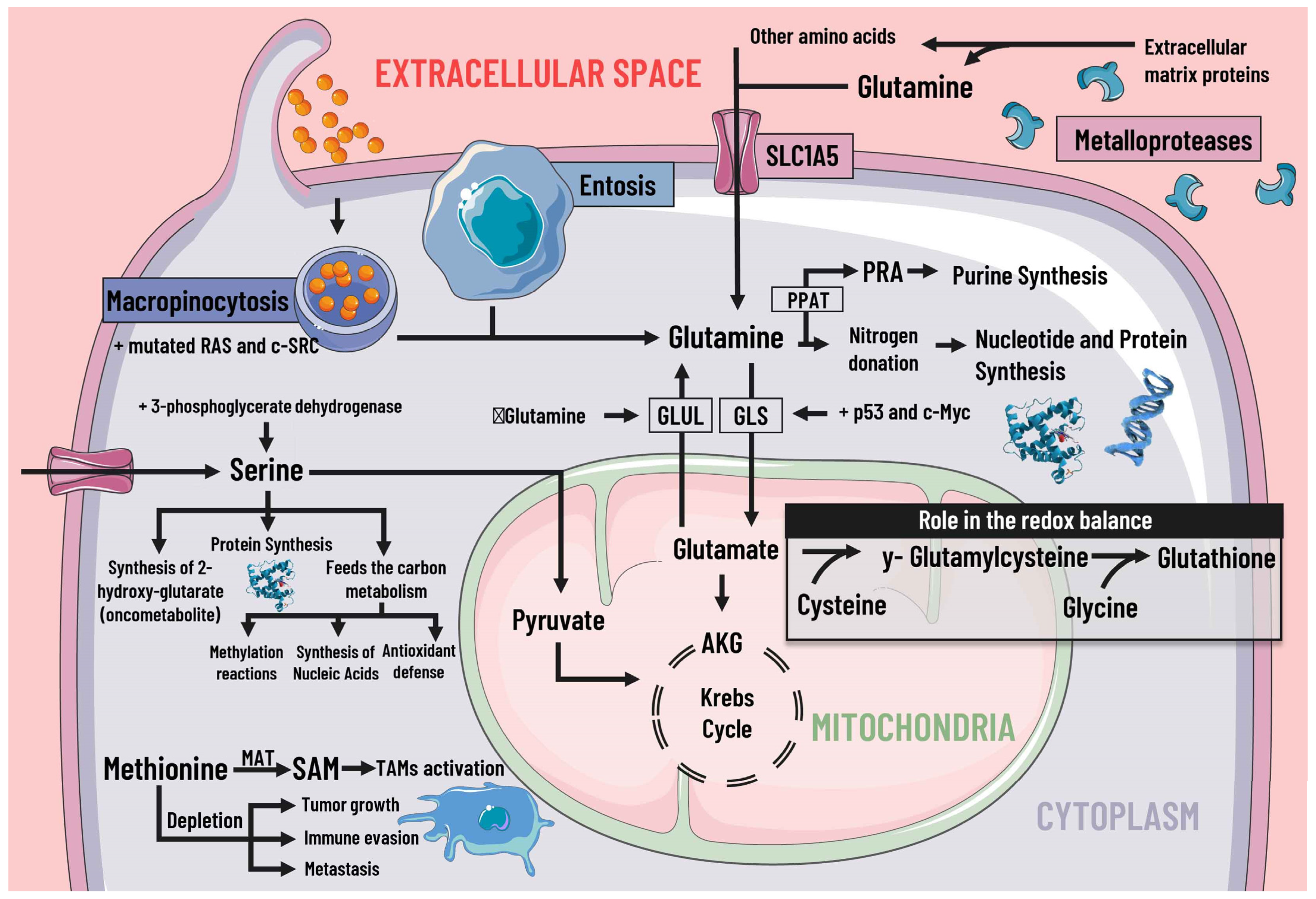
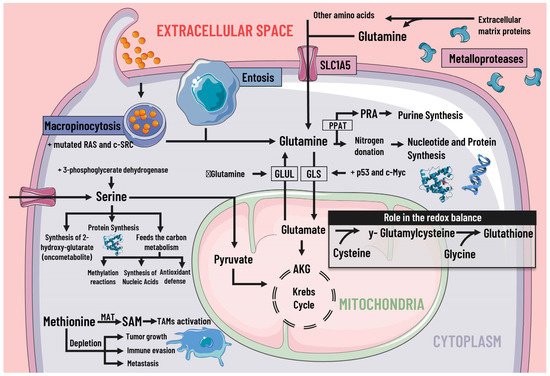
Figure 3. Protein metabolism and mediators of metabolic reprogramming. Protein metabolism in cancer cells is altered due to the immense demand generated by constant cell division, which is why amino acids are of great importance as proteogenic building blocks. Among the amino acids, glutamine is of great relevance in cancer cells. Glutamine enters the cell by different pathways, including macropinocytosis (which is increased by mutated RAS and c-SRC), entosis, degradation of cell-matrix proteins by metalloproteases, and transport by SLC1A5. Glutamine serves as a nitrogen donor and is used to synthesise nucleotides, purines, and proteins. It can also be a source of energy through glutaminolysis, which p53 and c-Myc stimulate. When there is a decrease in glutamine levels, it is synthesized by GLUL using glutamate as a substrate. Likewise, glutamine intervenes indirectly in the redox balance through glutamate. Serine is also essential, since increases in serine and 3-phosphoglycerate dehydrogenase, the first enzyme involved in its synthesis, are associated with tumor growth. Serine feeds carbon metabolism and protein synthesis, favors the synthesis of the oncometabolite 2-hydroxy-glutarate, and can act as an energy source, since it is an anaplerotic metabolite. In addition, methionine and SAM facilitate the pattern of histone methylation in monocytes/macrophages and the activation of TAMs. Depletion of exogenous methionine promotes tumor growth, metastasis, and immune evasion. SLC1A5 solute transporter family member 5; PPAT, phosphoribosyl pyrophosphate amidotransferase; PRA, phosphoribosylamine; GLS, glutaminase; GLUL, glutamine synthetase; AKG, alpha-ketoglutarate; SAMs S-adenosylmethionine; MAT, methionine adenosyltransferase; TAMs, tumor-associated macrophages.