Reprogramming is a process mediated by multiple factors, including oncogenes, growth factors, hypoxia-induced factors, and the loss of suppressor gene function, which support malignant transformation and tumor development in addition to cell heterogeneity. Consequently, this hallmark promotes resistance to conventional anti-tumor therapies by adapting to the drastic changes in the nutrient microenvironment that these therapies entail. Therefore, it represents a revolutionary landscape during cancer progression that could be useful for developing new and improved therapeutic strategies targeting alterations in cancer cell metabolism, such as the deregulated mTOR and PI3K pathways.
- metabolic reprogramming
- tumor microenvironment
- energy metabolism
1. Introduction
2. Metabolic Pathways in Cancer
2.1. Carbohydrate Metabolism
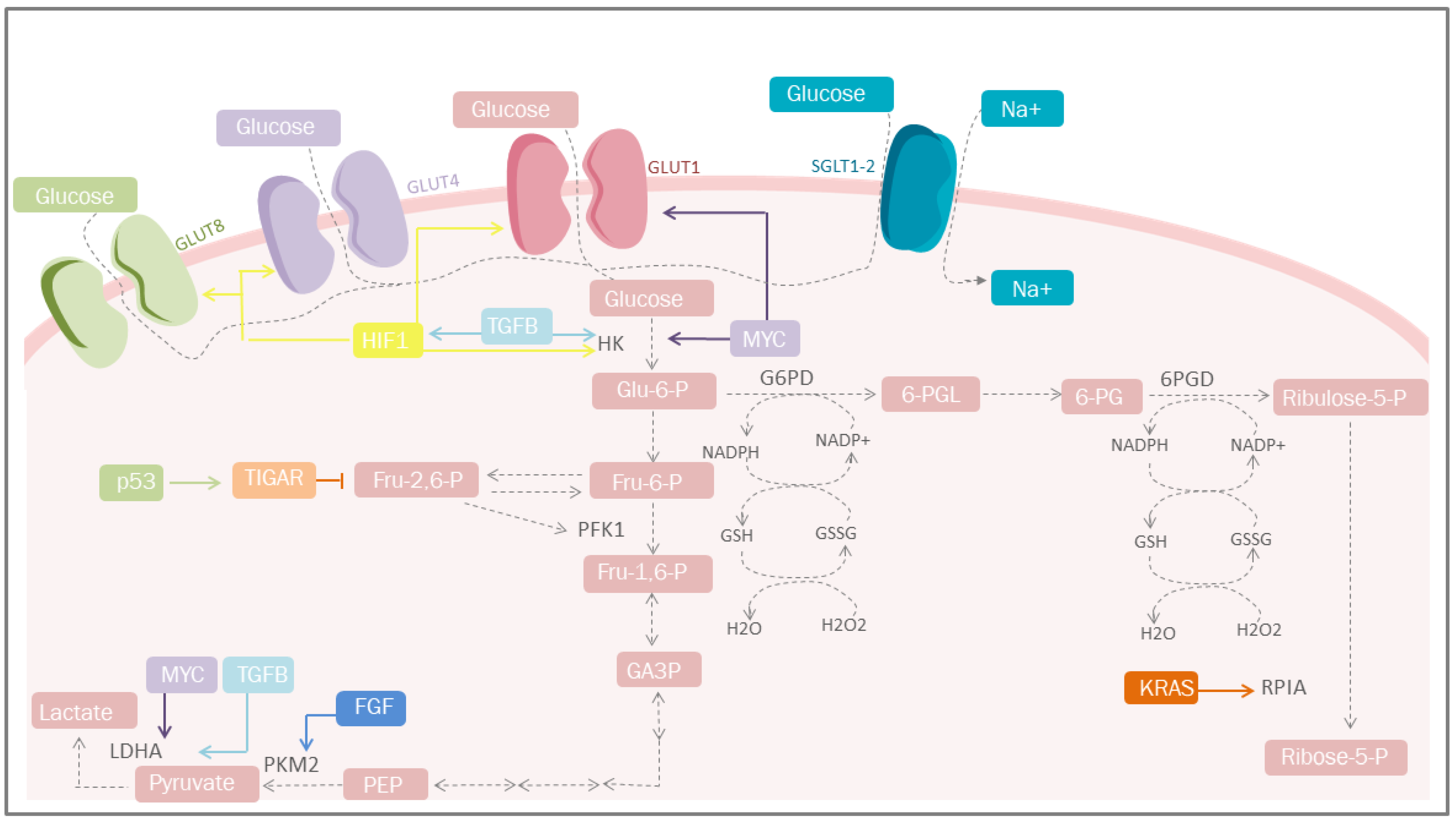 Figure 1. Carbohydrate metabolism and mediators of metabolic reprogramming. Cancer cells must acquire a greater amount of nutrients, especially glucose. One of the mediators is HIF-1, which increases glucose uptake through the induction of GLUT-1, GLUT-4 and GLUT-1; simultaneosly, it can also be stimulated by TGF-β through the PI3K/AKT/mTOR pathway. Other important mediators are p53 that plays a protective role against ROS. GLUT, glucose transporters; Glu-6-P, glucose 6-phosphate; Fru-6-P, fructose 6-bisphosphate; Fru-1,6-P, fructose 1,6-bisphosphate; Fru-2,6-P, fructose 2,6-bisphosphate; GA-3-P, glyceraldehyde 3-phosphate; PEP, phosphoenolpyruvate; HK, hexokinase; TGFB, transforming growth factor beta; PFK1, phosphofructokinase 1; PKM2, pyruvate kinase M2; LDHA, lactate dehydrogenase A; G6PD, glucose-6-phosphate dehydrogenase; 6-PGL, 6-phosphogluconolactonase; NADP+, nicotinamide adenine dinucleotide phosphate; NADPH, reduced form of NADP; GSSG, glutathione disulfide; GSH, glutathione; H2O2, hydrogen peroxide; TIGAR, Tp53-induced glycolysis and apoptosis regulator; FGF, fibroblast growth factor; HIF-1, hypoxia-inducible factor 1; PPP, pentose phosphate pathway; KRAS, Kirsten-ras; RPIA, ribose-5 phosphate isomerase; MYC, proto-oncogene; Ribulose-5-P, ribulose 5-phosphate; Ribose-5-P, ribose 5-phosphate; SGLT1/2, sodium-glucose cotransporter-1/2.
Figure 1. Carbohydrate metabolism and mediators of metabolic reprogramming. Cancer cells must acquire a greater amount of nutrients, especially glucose. One of the mediators is HIF-1, which increases glucose uptake through the induction of GLUT-1, GLUT-4 and GLUT-1; simultaneosly, it can also be stimulated by TGF-β through the PI3K/AKT/mTOR pathway. Other important mediators are p53 that plays a protective role against ROS. GLUT, glucose transporters; Glu-6-P, glucose 6-phosphate; Fru-6-P, fructose 6-bisphosphate; Fru-1,6-P, fructose 1,6-bisphosphate; Fru-2,6-P, fructose 2,6-bisphosphate; GA-3-P, glyceraldehyde 3-phosphate; PEP, phosphoenolpyruvate; HK, hexokinase; TGFB, transforming growth factor beta; PFK1, phosphofructokinase 1; PKM2, pyruvate kinase M2; LDHA, lactate dehydrogenase A; G6PD, glucose-6-phosphate dehydrogenase; 6-PGL, 6-phosphogluconolactonase; NADP+, nicotinamide adenine dinucleotide phosphate; NADPH, reduced form of NADP; GSSG, glutathione disulfide; GSH, glutathione; H2O2, hydrogen peroxide; TIGAR, Tp53-induced glycolysis and apoptosis regulator; FGF, fibroblast growth factor; HIF-1, hypoxia-inducible factor 1; PPP, pentose phosphate pathway; KRAS, Kirsten-ras; RPIA, ribose-5 phosphate isomerase; MYC, proto-oncogene; Ribulose-5-P, ribulose 5-phosphate; Ribose-5-P, ribose 5-phosphate; SGLT1/2, sodium-glucose cotransporter-1/2.2.2. Lipid Metabolism
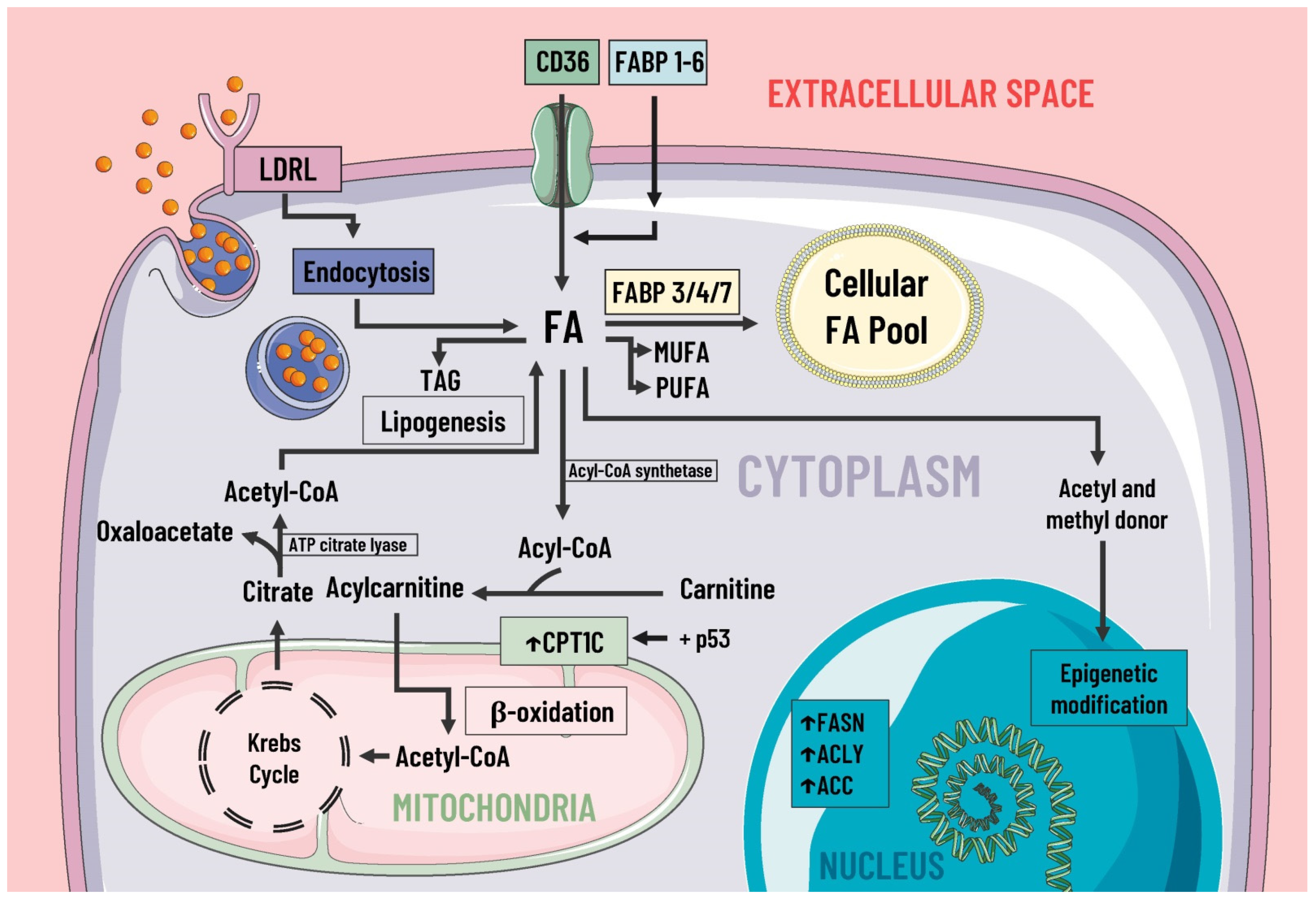 Figure 2. Lipids metabolism and mediators of metabolic reprogramming. Lipid metabolism in cancer cells is altered to increase the availability of these molecules, which provide structural components for the cell membrane, second messengers, and a fuel source. Fatty acids enter the cell by several pathways, including LDRL-mediated endocytosis and a wide variety of membrane transporters, such as CD36 and FABP1-6. Another source of FAS is lipogenesis. Once in the intracellular space, FAS can be stored in the cellular FAS pool by means of FABP 3/4/7, and can also serve as a substrate for the formation of MUFA, PUFA, TAG, etc. Likewise, some researchers suggest FAS are a great energy source through β-oxidation and act as donors of acetyl and methyl groups, participating in epigenetic modifications. In cancer, there is an increase in the expression of enzymes involved in lipogenesis, cholesterol synthesis, and beta-oxidation, such as ACC, FASN, ACLY, and CPT1C, where the latter is due to p53. LDRL, low-density lipoprotein receptor; CD36, a cluster of differentiation 36; FABP, fatty acid-binding protein; FAS, fatty acids synthetase; MUFA, monounsaturated fatty acids; PUFA, polyunsaturated fatty acids; TAG, triacylglycerides; CPT1C, carnitine palmitoyltransferase 1C; FASN, fatty acid synthase; ACLY, ATP citrate lyase; ACC, acetyl-CoA carboxylase; ↑, increase.
Figure 2. Lipids metabolism and mediators of metabolic reprogramming. Lipid metabolism in cancer cells is altered to increase the availability of these molecules, which provide structural components for the cell membrane, second messengers, and a fuel source. Fatty acids enter the cell by several pathways, including LDRL-mediated endocytosis and a wide variety of membrane transporters, such as CD36 and FABP1-6. Another source of FAS is lipogenesis. Once in the intracellular space, FAS can be stored in the cellular FAS pool by means of FABP 3/4/7, and can also serve as a substrate for the formation of MUFA, PUFA, TAG, etc. Likewise, some researchers suggest FAS are a great energy source through β-oxidation and act as donors of acetyl and methyl groups, participating in epigenetic modifications. In cancer, there is an increase in the expression of enzymes involved in lipogenesis, cholesterol synthesis, and beta-oxidation, such as ACC, FASN, ACLY, and CPT1C, where the latter is due to p53. LDRL, low-density lipoprotein receptor; CD36, a cluster of differentiation 36; FABP, fatty acid-binding protein; FAS, fatty acids synthetase; MUFA, monounsaturated fatty acids; PUFA, polyunsaturated fatty acids; TAG, triacylglycerides; CPT1C, carnitine palmitoyltransferase 1C; FASN, fatty acid synthase; ACLY, ATP citrate lyase; ACC, acetyl-CoA carboxylase; ↑, increase.2.3. Protein Metabolism
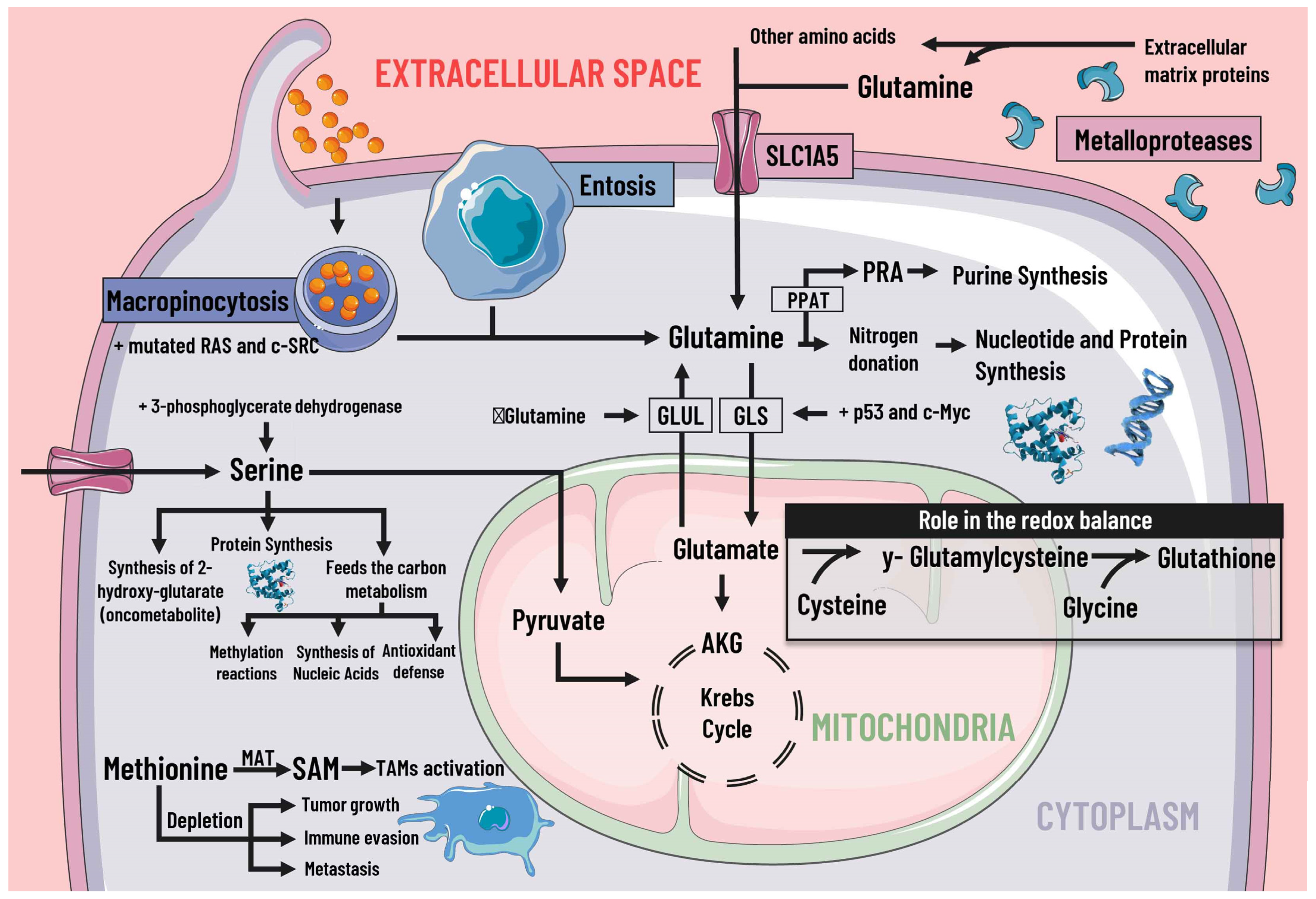 Figure 3. Protein metabolism and mediators of metabolic reprogramming. Protein metabolism in cancer cells is altered due to the immense demand generated by constant cell division, which is why amino acids are of great importance as proteogenic building blocks. Among the amino acids, glutamine is of great relevance in cancer cells. Glutamine enters the cell by different pathways, including macropinocytosis (which is increased by mutated RAS and c-SRC), entosis, degradation of cell-matrix proteins by metalloproteases, and transport by SLC1A5. Glutamine serves as a nitrogen donor and is used to synthesise nucleotides, purines, and proteins. It can also be a source of energy through glutaminolysis, which p53 and c-Myc stimulate. When there is a decrease in glutamine levels, it is synthesized by GLUL using glutamate as a substrate. Likewise, glutamine intervenes indirectly in the redox balance through glutamate. Serine is also essential, since increases in serine and 3-phosphoglycerate dehydrogenase, the first enzyme involved in its synthesis, are associated with tumor growth. Serine feeds carbon metabolism and protein synthesis, favors the synthesis of the oncometabolite 2-hydroxy-glutarate, and can act as an energy source, since it is an anaplerotic metabolite. In addition, methionine and SAM facilitate the pattern of histone methylation in monocytes/macrophages and the activation of TAMs. Depletion of exogenous methionine promotes tumor growth, metastasis, and immune evasion. SLC1A5 solute transporter family member 5; PPAT, phosphoribosyl pyrophosphate amidotransferase; PRA, phosphoribosylamine; GLS, glutaminase; GLUL, glutamine synthetase; AKG, alpha-ketoglutarate; SAMs S-adenosylmethionine; MAT, methionine adenosyltransferase; TAMs, tumor-associated macrophages.
Figure 3. Protein metabolism and mediators of metabolic reprogramming. Protein metabolism in cancer cells is altered due to the immense demand generated by constant cell division, which is why amino acids are of great importance as proteogenic building blocks. Among the amino acids, glutamine is of great relevance in cancer cells. Glutamine enters the cell by different pathways, including macropinocytosis (which is increased by mutated RAS and c-SRC), entosis, degradation of cell-matrix proteins by metalloproteases, and transport by SLC1A5. Glutamine serves as a nitrogen donor and is used to synthesise nucleotides, purines, and proteins. It can also be a source of energy through glutaminolysis, which p53 and c-Myc stimulate. When there is a decrease in glutamine levels, it is synthesized by GLUL using glutamate as a substrate. Likewise, glutamine intervenes indirectly in the redox balance through glutamate. Serine is also essential, since increases in serine and 3-phosphoglycerate dehydrogenase, the first enzyme involved in its synthesis, are associated with tumor growth. Serine feeds carbon metabolism and protein synthesis, favors the synthesis of the oncometabolite 2-hydroxy-glutarate, and can act as an energy source, since it is an anaplerotic metabolite. In addition, methionine and SAM facilitate the pattern of histone methylation in monocytes/macrophages and the activation of TAMs. Depletion of exogenous methionine promotes tumor growth, metastasis, and immune evasion. SLC1A5 solute transporter family member 5; PPAT, phosphoribosyl pyrophosphate amidotransferase; PRA, phosphoribosylamine; GLS, glutaminase; GLUL, glutamine synthetase; AKG, alpha-ketoglutarate; SAMs S-adenosylmethionine; MAT, methionine adenosyltransferase; TAMs, tumor-associated macrophages.2.4. Methionine, S-Adenosylmethionine and Cysteine
2.5. Isoenzymes
2.5.1. Fumarate Hydratase (FH)
2.5.2. Isocitrate Dehydrogenase
2.5.3. Glutaminase
2.5.4. Lactate Dehydrogenases
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics14061303
References
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.K.; Ouellette, B.F.F.; Li, C.H.; et al. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93.
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer Statistics for the Year 2020: An Overview. Int. J. Cancer 2021, 149, 778–789.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674.
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46.
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200.
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218.
- da Silva-Diz, V.; Lorenzo-Sanz, L.; Bernat-Peguera, A.; Lopez-Cerda, M.; Muñoz, P. Cancer Cell Plasticity: Impact on Tumor Progression and Therapy Response. Semin. Cancer Biol. 2018, 53, 48–58.
- Lue, H.; Podolak, J.; Kolahi, K.; Cheng, L.; Rao, S.; Garg, D.; Xue, C.-H.; Rantala, J.K.; Tyner, J.W.; Thornburg, K.L.; et al. Metabolic Reprogramming Ensures Cancer Cell Survival despite Oncogenic Signaling Blockade. Genes Dev. 2017, 31, 2067–2084.
- Sun, L.; Suo, C.; Li, S.-T.; Zhang, H.; Gao, P. Metabolic Reprogramming for Cancer Cells and Their Microenvironment: Beyond the Warburg Effect. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 51–66.
- Xia, L.; Oyang, L.; Lin, J.; Tan, S.; Han, Y.; Wu, N.; Yi, P.; Tang, L.; Pan, Q.; Rao, S.; et al. The Cancer Metabolic Reprogramming and Immune Response. Mol. Cancer 2021, 20, 28.
- Li, T.; Tan, X.; Yang, R.; Miao, Y.; Zhang, M.; Xi, Y.; Guo, R.; Zheng, M.; Li, B. Discovery of Novel Glyceraldehyde-3-Phosphate Dehydrogenase Inhibitor via Docking-Based Virtual Screening. Bioorg. Chem. 2020, 96, 103620.
- Ghanbari Movahed, Z.; Rastegari-Pouyani, M.; Mohammadi, M.H.; Mansouri, K. Cancer Cells Change Their Glucose Metabolism to Overcome Increased ROS: One Step from Cancer Cell to Cancer Stem Cell? Biomed. Pharmacother. 2019, 112, 108690.
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47.
- Sun, R.C.; Dukhande, V.V.; Zhou, Z.; Young, L.E.A.; Emanuelle, S.; Brainson, C.F.; Gentry, M.S. Nuclear Glycogenolysis Modulates Histone Acetylation in Human Non-Small Cell Lung Cancers. Cell Metab. 2019, 30, 903–916.
- Grasmann, G.; Smolle, E.; Olschewski, H.; Leithner, K. Gluconeogenesis in Cancer Cells—Repurposing of a Starvation-Induced Metabolic Pathway? Biochim. Biophys. Acta BBA Rev. Cancer 2019, 1872, 24–36.
- Vincent, E.E.; Sergushichev, A.; Griss, T.; Gingras, M.-C.; Samborska, B.; Ntimbane, T.; Coelho, P.P.; Blagih, J.; Raissi, T.C.; Choinière, L.; et al. Mitochondrial Phosphoenolpyruvate Carboxykinase Regulates Metabolic Adaptation and Enables Glucose-Independent Tumor Growth. Mol. Cell 2015, 60, 195–207.
- Zhou, L.; Luo, M.; Cheng, L.-J.; Li, R.-N.; Liu, B.; Linghu, H. Glutamine-Fructose-6-Phosphate Transaminase 2 (GFPT2) Promotes the EMT of Serous Ovarian Cancer by Activating the Hexosamine Biosynthetic Pathway to Increase the Nuclear Location of β-Catenin. Pathol. Res. Pract. 2019, 215, 152681.
- Bu, P.; Chen, K.Y.; Xiang, K.; Johnson, C.; Crown, S.B.; Rakhilin, N.; Ai, Y.; Wang, L.; Xi, R.; Astapova, I.; et al. Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell Metab. 2018, 27, 1249–1262.
- Hammond, G.R.V.; Burke, J.E. Novel Roles of Phosphoinositides in Signaling, Lipid Transport, and Disease. Curr. Opin. Cell Biol. 2020, 63, 57–67.
- Fernández, L.P.; Gómez de Cedrón, M.; Ramírez de Molina, A. Alterations of Lipid Metabolism in Cancer: Implications in Prognosis and Treatment. Front. Oncol. 2020, 10, 577420.
- Wang, J.; Li, Y. CD36 Tango in Cancer: Signaling Pathways and Functions. Theranostics 2019, 9, 4893–4908.
- Tanase, C.; Enciu, A.M.; Codrici, E.; Popescu, I.D.; Dudau, M.; Dobri, A.M.; Pop, S.; Mihai, S.; Gheorghișan-Gălățeanu, A.-A.; Hinescu, M.E. Fatty Acids, CD36, Thrombospondin-1, and CD47 in Glioblastoma: Together and/or Separately? Int. J. Mol. Sci. 2022, 23, 604.
- Hu, J.; Zhang, L.; Chen, W.; Shen, L.; Jiang, J.; Sun, S.; Chen, Z. Role of Intra- and Extracellular Lipid Signals in Cancer Stemness and Potential Therapeutic Strategy. Front. Pharmacol. 2021, 12, 730751.
- Fhu, C.W.; Ali, A. Fatty Acid Synthase: An Emerging Target in Cancer. Molecules 2020, 25, 3935.
- Castelli, S.; De Falco, P.; Ciccarone, F.; Desideri, E.; Ciriolo, M.R. Lipid Catabolism and ROS in Cancer: A Bidirectional Liaison. Cancers 2021, 13, 5484.
- Bartolacci, C.; Andreani, C.; El-Gammal, Y.; Scaglioni, P.P. Lipid Metabolism Regulates Oxidative Stress and Ferroptosis in RAS-Driven Cancers: A Perspective on Cancer Progression and Therapy. Front. Mol. Biosci. 2021, 8, 706650.
- Casals, N.; Zammit, V.; Herrero, L.; Fadó, R.; Rodríguez-Rodríguez, R.; Serra, D. Carnitine Palmitoyltransferase 1C: From Cognition to Cancer. Prog. Lipid Res. 2016, 61, 134–148.
- Parrales, A.; Iwakuma, T. P53 as a Regulator of Lipid Metabolism in Cancer. Int. J. Mol. Sci. 2016, 17, 2074.
- Bott, A.J.; Shen, J.; Tonelli, C.; Zhan, L.; Sivaram, N.; Jiang, Y.-P.; Yu, X.; Bhatt, V.; Chiles, E.; Zhong, H.; et al. Glutamine Anabolism Plays a Critical Role in Pancreatic Cancer by Coupling Carbon and Nitrogen Metabolism. Cell Rep. 2019, 29, 1287–1298.
- Vettore, L.; Westbrook, R.L.; Tennant, D.A. New Aspects of Amino Acid Metabolism in Cancer. Br. J. Cancer 2020, 122, 150–156.
- Hosios, A.M.; Hecht, V.C.; Danai, L.V.; Johnson, M.O.; Rathmell, J.C.; Steinhauser, M.L.; Manalis, S.R.; Vander Heiden, M.G. Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev. Cell 2016, 36, 540–549.
- Kurbegovic, A.; Trudel, M. The Master Regulators Myc and P53 Cellular Signaling and Functions in Polycystic Kidney Disease. Cell. Signal. 2020, 71, 109594.
- Spinelli, J.B.; Yoon, H.; Ringel, A.E.; Jeanfavre, S.; Clish, C.B.; Haigis, M.C. Metabolic Recycling of Ammonia via Glutamate Dehydrogenase Supports Breast Cancer Biomass. Science 2017, 358, 941–946.
- Lukey, M.J.; Katt, W.P.; Cerione, R.A. Targeting Amino Acid Metabolism for Cancer Therapy. Drug Discov. Today 2017, 22, 796–804.
- Corchado-Cobos, R.; García-Sancha, N.; Mendiburu-Eliçabe, M.; Gómez-Vecino, A.; Jiménez-Navas, A.; Pérez-Baena, M.J.; Holgado-Madruga, M.; Mao, J.-H.; Cañueto, J.; Castillo-Lluva, S.; et al. Pathophysiological Integration of Metabolic Reprogramming in Breast Cancer. Cancers 2022, 14, 322.
- Durgan, J.; Florey, O. Cancer Cell Cannibalism: Multiple Triggers Emerge for Entosis. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 831–841.
- Zhang, Y.; Yang, H.; Zhao, J.; Wan, P.; Hu, Y.; Lv, K.; Hu, Y.; Yang, X.; Ma, M. Activation of MAT2A-RIP1 Signaling Axis Reprograms Monocytes in Gastric Cancer. J. Immunother. Cancer 2021, 9, e001364.
- Zhang, H.-F.; Klein Geltink, R.I.; Parker, S.J.; Sorensen, P.H. Transsulfuration, Minor Player or Crucial for Cysteine Homeostasis in Cancer. Trends Cell Biol. 2022, S0962-8924(22)000605.
- Tyrakis, P.A.; Yurkovich, M.E.; Sciacovelli, M.; Papachristou, E.K.; Bridges, H.R.; Gaude, E.; Schreiner, A.; D’Santos, C.; Hirst, J.; Hernandez-Fernaud, J.; et al. Fumarate Hydratase Loss Causes Combined Respiratory Chain Defects. Cell Rep. 2017, 21, 1036–1047.
- Gonçalves, E.; Sciacovelli, M.; Costa, A.S.H.; Tran, M.G.B.; Johnson, T.I.; Machado, D.; Frezza, C.; Saez-Rodriguez, J. Post-Translational Regulation of Metabolism in Fumarate Hydratase Deficient Cancer Cells. Metab. Eng. 2018, 45, 149–157.
- Schmidt, S.; Gay, D.; Uthe, F.W.; Denk, S.; Paauwe, M.; Matthes, N.; Diefenbacher, M.E.; Bryson, S.; Warrander, F.C.; Erhard, F.; et al. A MYC–GCN2–EIF2α Negative Feedback Loop Limits Protein Synthesis to Prevent MYC-Dependent Apoptosis in Colorectal Cancer. Nat. Cell Biol. 2019, 21, 1413–1424.
- Masui, K.; Onizuka, H.; Cavenee, W.K.; Mischel, P.S.; Shibata, N. Metabolic Reprogramming in the Pathogenesis of Glioma: Update. Neuropathol. Off. J. Jpn. Soc. Neuropathol. 2019, 39, 3–13.
- Gelman, S.J.; Naser, F.; Mahieu, N.G.; McKenzie, L.D.; Dunn, G.P.; Chheda, M.G.; Patti, G.J. Consumption of NADPH for 2-HG Synthesis Increases Pentose Phosphate Pathway Flux and Sensitizes Cells to Oxidative Stress. Cell Rep. 2018, 22, 512–522.
- Izquierdo-Garcia, J.L.; Viswanath, P.; Eriksson, P.; Cai, L.; Radoul, M.; Chaumeil, M.M.; Blough, M.; Luchman, H.A.; Weiss, S.; Cairncross, J.G.; et al. IDH1 Mutation Induces Reprogramming of Pyruvate Metabolism. Cancer Res. 2015, 75, 2999–3009.
- Fitzpatrick, S.F.; Lambden, S.; Macias, D.; Puthucheary, Z.; Pietsch, S.; Mendil, L.; McPhail, M.J.W.; Johnson, R.S. 2-Hydroxyglutarate Metabolism Is Altered in an In Vivo Model of LPS Induced Endotoxemia. Front. Physiol. 2020, 11, 1–8.
- Jiang, J.; Srivastava, S.; Zhang, J. Starve Cancer Cells of Glutamine: Break the Spell or Make a Hungry Monster? Cancers 2019, 11, 804.
- Masisi, B.K.; El Ansari, R.; Alfarsi, L.; Rakha, E.A.; Green, A.R.; Craze, M.L. The Role of Glutaminase in Cancer. Histopathology 2020, 76, 498–508.
- Katt, W.P.; Lukey, M.J.; Cerione, R.A. A Tale of Two Glutaminases: Homologous Enzymes with Distinct Roles in Tumorigenesis. Future Med. Chem. 2017, 9, 223–243.
- Matés, J.M.; Campos-Sandoval, J.A.; Márquez, J. Glutaminase Isoenzymes in the Metabolic Therapy of Cancer. Biochim. Biophys. Acta BBA Rev. Cancer 2018, 1870, 158–164.
- Gouirand, V.; Guillaumond, F.; Vasseur, S. Influence of the Tumor Microenvironment on Cancer Cells Metabolic Reprogramming. Front. Oncol. 2018, 8, 117.