Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Donat Kögel | -- | 1908 | 2022-06-15 10:09:01 | | | |
| 2 | Peter Tang | -4 word(s) | 1904 | 2022-06-15 11:40:57 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kögel, D.; Linder, B. Autophagy in Cancer Cell Death. Encyclopedia. Available online: https://encyclopedia.pub/entry/24052 (accessed on 02 August 2026).
Kögel D, Linder B. Autophagy in Cancer Cell Death. Encyclopedia. Available at: https://encyclopedia.pub/entry/24052. Accessed August 02, 2026.
Kögel, Donat, Benedikt Linder. "Autophagy in Cancer Cell Death" Encyclopedia, https://encyclopedia.pub/entry/24052 (accessed August 02, 2026).
Kögel, D., & Linder, B. (2022, June 15). Autophagy in Cancer Cell Death. In Encyclopedia. https://encyclopedia.pub/entry/24052
Kögel, Donat and Benedikt Linder. "Autophagy in Cancer Cell Death." Encyclopedia. Web. 15 June, 2022.
Copy Citation
Autophagy has important functions in maintaining energy metabolism under conditions of starvation and to alleviate stress by removal of damaged and potentially harmful cellular components. Therefore, autophagy represents a pro-survival stress response in the majority of cases. An alternative pro-death function of autophagy has been consistently observed in different settings, in particular, in developmental cell death of lower organisms and in drug-induced cancer cell death. This cell death is referred to as autophagic cell death (ACD) or autophagy-dependent cell death (ADCD), a type of cellular demise that may act as a backup cell death program in apoptosis-deficient tumors.
autophagy
cell death
cancer
gossypol
AT-101
mitophagy
mitochondria
1. Autophagy in Cell Survival and Cell Death
One such apoptosis-independent cell death mechanism relies on the over activation of autophagy, a cellular stress response that normally serves as a quality control mechanism. Different forms of autophagy have been described including macroautophagy (hereafter referred to as autophagy), microautophagy, and chaperone-mediated autophagy. The molecular basis of mammalian autophagy is depicted in Figure 1.
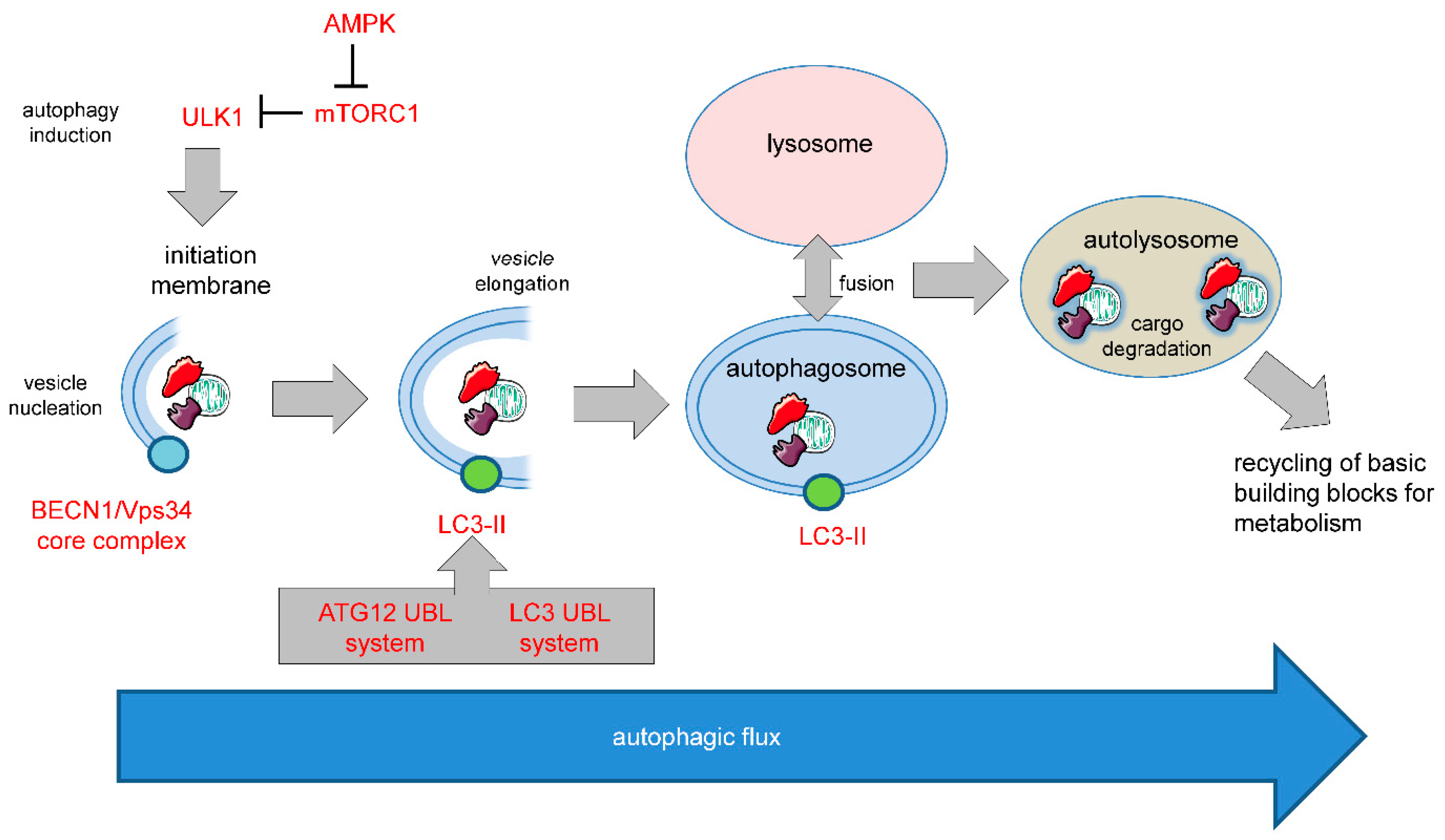
Figure 1. Molecular basis of mammalian autophagy. Autophagy is a multistep process involving several key ATG proteins and signaling complexes. It requires the formation of double-membrane-containing autophagosomes that sequester proteins, lipids, organelles or invasive microbes and fuse with lysosomes for digestion of content by acidic hydrolases. ULK1, a protein kinase serving as the central initiator of autophagy, is inhibited by the mTORC1 complex that contains mTOR. AMPK serves as a nutrient sensor and negative regulator of mTORC1. Autophagosome biogenesis starts with the formation of an initiation membrane that is derived either from the endoplasmatic reticulum (ER) or from several other cellular membrane sources. Vesicle nucleation is promoted by the BECN1/Vps34 core complex containing the lipid kinase Vps34. Vesicle elongation is regulated by the two ubiquitin-like conjugation systems (UBLs) ATG12-UBL and LC3-UBL that cooperate to catalyze the conjugation of phosphatidylethanolamine (PE) to LC3 and facilitate the conversion of cytosolic LC3-I into a membrane-associated LC3-II that is translocated to the autophagosomal membrane. Following vesicle closure, mature autophagosomes fuse with lysosomes to generate autolysosomes that digest the autophagosomal content by lysosomal proteases for cellular recycling [1]. This figure was created using Servier Medical Art templates, which are licensed under a Creative Commons Attribution 3.0 Unported License; https://smart.servier.com.
In general, autophagy is a pro-survival stress response, for example, autophagy will be activated under situations of nutrient deprivation to ensure supply of basic building blocks for metabolism and survival of the cells/organisms by recycling of non-essential cellular components. Autophagy also serves to remove damaged and potentially harmful organelles, thereby supporting cell survival. On the other hand, there is conclusive evidence that prolonged over activation of the autophagosomal/lysosomal pathway can lead to autophagic cell death (ACD, type II cell death). Of note, similar threshold effects on cell survival vs cell death are commonly observed in various stress responses like the endoplasmatic reticulum (ER) stress response and activation of p53 [2]. Accordingly, ACD is often described as self-digestion beyond the point allowing cell survival [2][3][4][5][6]. Hence, the net effect of autophagy on cell survival is highly dependent on its intensity and duration, but also on its particular context (Figure 2).
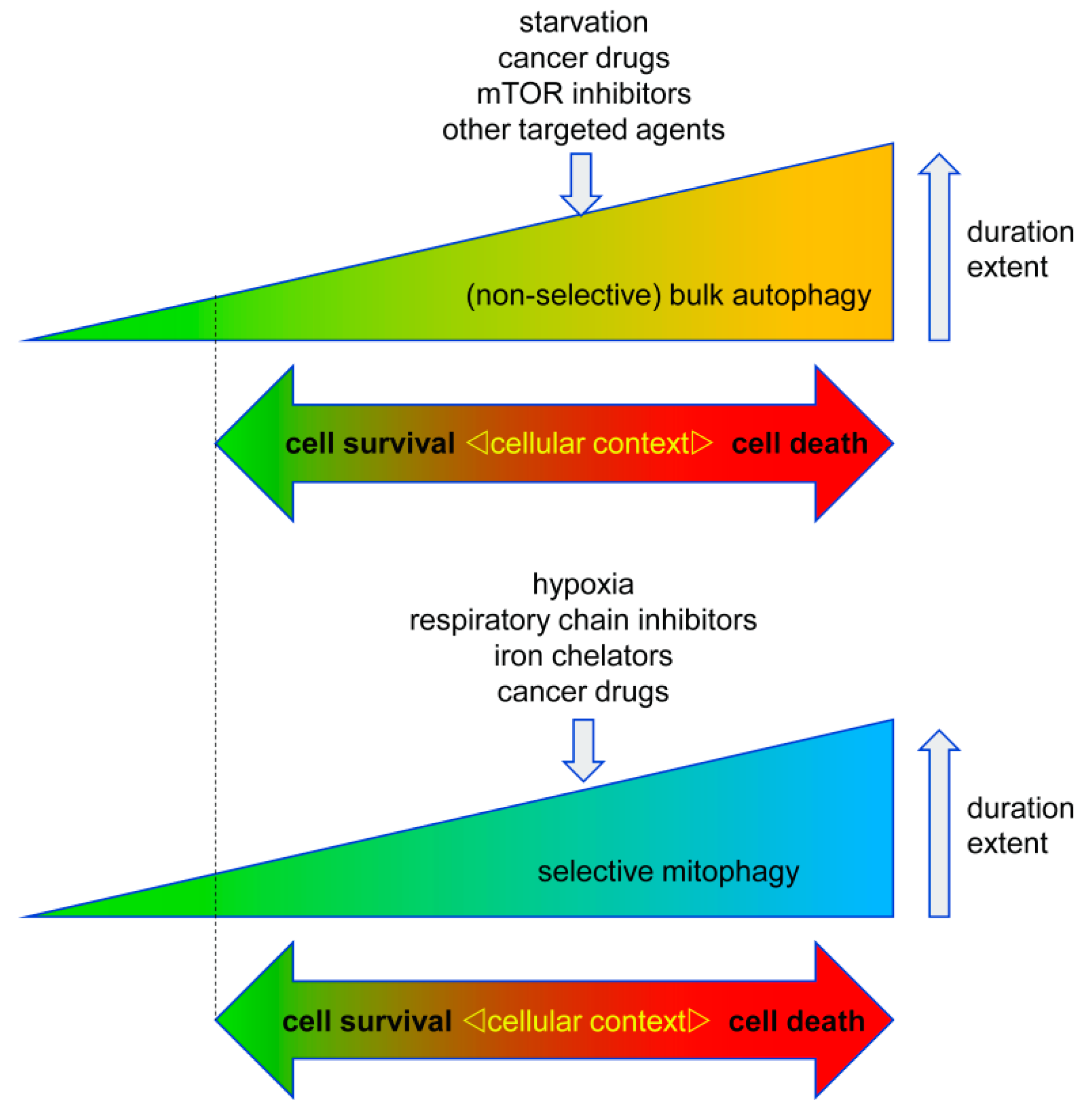
Figure 2. Context-dependent cell responses to bulk autophagy and selective mitophagy. Both autophagy and mitophagy can either promote or inhibit cell death in cancer cells. This response is highly dependent on the cell type, the trigger of auto-/mitophagy, its duration, and its extent. Accordingly, excessive autophagy can lead to cell death, however too little autophagy/mitophagy (marked by dotted line) can also be detrimental to the cells due to an impaired quality control/removal of harmful cellular material.
2. Autophagy in Tumorigenesis and Tumor Progression
Autophagy is not only involved in the responses to tumor therapy (see below), but also plays a key role in cancer development. This involvement of autophagy in tumorigenesis and tumor progression is very complex and multifaceted. It is hypothesized that in the early stages of tumorigenesis, autophagy exerts a tumor-suppressive function. This is based on the observation that an intact autophagy pathway correlates with decreased oxidative stress and increased genomic stability [6], thereby ensuring the survival of healthy, non-transformed cells. On the other hand, it was recently proposed that autophagy may also support cancer progression by facilitating tumor cell survival and fitness under replication stress, a common feature of most malignancies [7].
In line with the high context-dependency of autophagy, there is a shift from tumor-suppressing to tumor-promoting autophagy during the course of tumor progression. It is believed that autophagy can alleviate the stressful environmental conditions like hypoxia or nutrient deprivation often encountered in manifest, solid tumors [8][9], but also in non-malignant ischemic tissues [10], rendering the tumors more stress-resistant. Using a Drosophila model, Katheder et al. recently showed that not only tumor-intrinsic but also microenvironmental autophagy is capable of inducing tumor growth by providing the nutrients necessary for tumor growth [11]. They could further demonstrate that this was achieved through elevated ROS levels due to mitochondrial damage in the tumor cells, which induced nutrient export from the microenvironment.
Since another key hallmark of cancer is chronic inflammation, it will also be of key importance to better understand the mutual interplay between autophagy and inflammation. There is evidence suggesting that autophagy can either suppress or promote inflammation in cancer. Likewise, inflammatory pathways can either suppress or induce autophagy in a context-dependent manner. A complex scenario is recently emerging that will aid future studies aimed at deciphering the exact role of autophagy in shaping the immune and inflammatory microenvironment of tumors [12].
In addition to supporting tumor growth in general, autophagy has been demonstrated to regulate and/or maintain the cancer stem cell phenotype and treatment resistance in multiple studies, for example, in oral squamous cell carcinoma [13] and endometrial cancer [14].
3. Autophagy in Therapy Response
Next to its role in tumorigenesis and malignant progression, autophagy plays a key role in cancer therapy responses. Given the dual function of autophagy in cell survival vs cell death, inhibition, but also over activation of autophagy, carries potential relevance for therapy.
3.1. Pro-Survival Autophagy
Since autophagy appears to act mainly as a pro-survival stress response that is activated (at least to some degree) by most, if not all, conventional cancer drugs and by radiation, pro-survival autophagy is expected to hamper the effects of cancer therapy in most settings. Some examples of a therapy resistance-increasing effect of autophagy are listed below. The impact of pro-survival autophagy in cancer therapy was extensively covered elsewhere in [15] where the researchers also delineated the molecular mechanisms of autophagy regulation in response to therapy-related stress conditions in this context, and the researchers refer the reader to this work for further details. Two central cellular players involved in many paradigms of pro-survival autophagy of cancer cells are mTOR and AMPK (Figure 1) that are often involved in activation of autophagy as an unwanted side effect of different cancer drugs/treatments. For example, treatment with Taxol was shown to activate pro-survival autophagy caused by inhibition of mTOR in breast cancer cells [16].
For pancreatic cancer, it has been shown that primary tumors and cell lines exhibit increased autophagy, while autophagy inhibition (genetic and pharmacological) results in increased reactive oxygen species (ROS) formation and DNA damage, while treatment of tumor-bearing mice with the autophagic flux-inhibitor chloroquine (CQ) improved overall survival [17]. In another study Qiu et al. showed that autophagy induced by cisplatin protected ovarian cancer cells [18], while DeVorkin et al. could, in fact, show that cancer cells of clear-cell ovarian cancer depend on autophagy for their survival [19]. From a mechanistic perspective, it should, however, be noted that CQ is not a highly selective inhibitor of autophagy. A recent study demonstrated that CQ also has profound non-autophagic effects on cells, especially concerning disorganization of the Golgi and endo-lysosomal systems [20], arguing for a more cautious interpretation of responses to CQ.
The approach of combining conventional or targeted therapy with autophagy inhibition (CQ, Hydroxy-CQ) is currently also investigated in several clinical studies in patients with various types of cancer, including glioblastoma. Accordingly, Jutten et al. showed recently that glioma cells expressing mutant EGFRvIII that is associated with poor prognosis [21] and occurs in half of all glioblastoma patients [22], are more sensitive to CQ treatment, and hence rely more strongly on autophagy for cell survival. Most importantly, using a retrospective analysis, this study also showed that patients with mutant EGFRvIII receiving CQ have the highest benefit of CQ-treated patients [23]. Another recent, very promising study provided evidence that autophagy inhibition can be employed to overcome therapy resistance of brain tumor patients against BRAF inhibitor treatment [24].
3.2. Pro-Death Autophagy
Given the fact that genetic and pharmacological abrogation of autophagy inhibits non-selective as well as selective types of autophagy, it is currently not well understood whether excessive pro-death bulk autophagy, i.e., non-selective autophagy, is the (solely) responsible type of autophagy for cell killing in most established paradigms of ACD, including ACD in lower organisms and ACD induced by cancer drugs. The following section lists several examples from the literature that lack evidence for a death-promoting contribution of selective autophagy pathways, such as mitophagy (see next paragraph).
Resveratrol, a polyphenolic compound found in red wine [25], has been described to induce bona fide ACD in chronic myeloid leukemia [26] and induces cell death in prostate [27], ovarian [28], and endometrial cancer cells [29] that involves induction of autophagy, although the latter studies failed to provide complete evidence that the criteria required by the NCCD [3] are fulfilled. A recent shRNA-based screen of A549 lung cancer cells analyzed potential regulators of resveratrol-induced ACD and identified glucosylceramidase beta (GBA1) as a potential mediator of ACD [30]. ACD has also been observed in cells treated with Interferon-gamma (IFN-γ) which induced cell death that could be rescued after treatment with the autophagy-inhibitor 3-methyl-adenine (3MA) or knockdown of ATG5 [31]. Based on the observations that cancer cells have a higher turnover rate of NAD+, this pathway was recently employed to target cancer cells by triggering ACD via inhibition of the NAD+-synthesizing enzyme Nampt using the inhibitor FK866 in myeloma [32] or by inhibition of the nicotinamide phosphoribosyltransferase by APO866 in leukemia and lymphoma cells [33]. Lima et al. used SK1-I, an inhibitor of sphingosine kinase 1 (SPHK1) and analog of sphingosine, in colon cancer cell lines and observed induction of autophagy and cell death which was dependent on BECN1 and ATG5 [34], although in this study the discrimination between apoptosis and autophagy is not entirely clear, leaving some room for interpretation if the mode of death can be truly defined as ACD according to the NCCD criteria. Other groups showed that downregulation of the AKT1/mTOR-axis using the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) induced ACD in hepatocellular carcinoma (HCC) cell lines [35]. Finally, the cholesterol metabolite dendrogenin A (DDA) induced lethal autophagy, reminiscent of ACD, in myeloma and acute myeloid leukemia in vitro and in vivo [36].
Arsenic trioxide was shown to induce ACD and cell death in various tumor cell populations in multiple studies [37][38][39][40]. Considering that arsenic trioxide is already clinically used to treat acute promyelocytic leukemia (APL) [41] and easily crosses the blood-brain-barrier [42], this drug could be particularly interesting for hard-to-treat cancers, such as brain tumors (primary or metastases). In particular, it was shown that arsenic trioxide-induced ACD is mediated by the protein BNIP3 (BCL2 interacting protein 3) and BNIP3L (BCL2 interacting protein 3 like; also known as NIX) [37], that were subsequently identified as mitophagy-receptors [43]. These findings imply that arsenic trioxide triggers selective autophagy of mitochondria (mitophagy) in addition to non-selective bulk-autophagy, with possible implications for cell death activation. However, this proposition warrants future research.
References
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell. Biol. 2014, 15, 81–94.
- Munoz-Pinedo, C.; Martin, S.J. Autosis: A new addition to the cell death Tower of Babel. Cell Death Dis. 2014, 5, e1319.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541.
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459.
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12 (Suppl. 2), 1509–1518.
- Gozuacik, D.; Kimchi, A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004, 23, 2891–2906.
- Vanzo, R.; Bartkova, J.; Merchut-Maya, J.M.; Hall, A.; Bouchal, J.; Dyrskjot, L.; Frankel, L.B.; Gorgoulis, V.; Maya-Mendoza, A.; Jaattela, M.; et al. Autophagy role(s) in response to oncogenes and DNA replication stress. Cell Death Differ. 2019.
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64.
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410.
- Hamacher-Brady, A.; Brady, N.R.; Gottlieb, R.A. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J. Biol. Chem. 2006, 281, 29776–29787.
- Katheder, N.S.; Khezri, R.; O’Farrell, F.; Schultz, S.W.; Jain, A.; Rahman, M.M.; Schink, K.O.; Theodossiou, T.A.; Johansen, T.; Juhasz, G.; et al. Microenvironmental autophagy promotes tumour growth. Nature 2017, 541, 417–420.
- Monkkonen, T.; Debnath, J. Inflammatory signaling cascades and autophagy in cancer. Autophagy 2018, 14, 190–198.
- Naik, P.P.; Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, C.K.; Mishra, R.; Patil, S.; Bhutia, S.K. Autophagy regulates cisplatin-induced stemness and chemoresistance via the upregulation of CD44, ABCB1 and ADAM17 in oral squamous cell carcinoma. Cell Prolif. 2018, 51, e12411.
- Ran, X.; Zhou, P.; Zhang, K. Autophagy plays an important role in stemness mediation and the novel dual function of EIG121 in both autophagy and stemness regulation of endometrial carcinoma JEC cells. Int. J. Oncol. 2017, 51, 644–656.
- Das, C.K.; Mandal, M.; Kogel, D. Pro-survival autophagy and cancer cell resistance to therapy. Cancer Metastasis Rev. 2018, 37, 749–766.
- Notte, A.; Ninane, N.; Arnould, T.; Michiels, C. Hypoxia counteracts taxol-induced apoptosis in MDA-MB-231 breast cancer cells: Role of autophagy and JNK activation. Cell Death Dis. 2013, 4, e638.
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729.
- Qiu, S.; Sun, L.; Jin, Y.; An, Q.; Weng, C.; Zheng, J. Silencing of BAG3 promotes the sensitivity of ovarian cancer cells to cisplatin via inhibition of autophagy. Oncol. Rep 2017, 38, 309–316.
- DeVorkin, L.; Hattersley, M.; Kim, P.; Ries, J.; Spowart, J.; Anglesio, M.S.; Levi, S.M.; Huntsman, D.G.; Amaravadi, R.K.; Winkler, J.D.; et al. Autophagy Inhibition Enhances Sunitinib Efficacy in Clear Cell Ovarian Carcinoma. Mol. Cancer Res. 2017, 15, 250–258.
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455.
- Pelloski, C.E.; Ballman, K.V.; Furth, A.F.; Zhang, L.; Lin, E.; Sulman, E.P.; Bhat, K.; McDonald, J.M.; Yung, W.K.; Colman, H.; et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J. Clin. Oncol. 2007, 25, 2288–2294.
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009, 16, 748–754.
- Jutten, B.; Keulers, T.G.; Peeters, H.J.M.; Schaaf, M.B.E.; Savelkouls, K.G.M.; Compter, I.; Clarijs, R.; Schijns, O.; Ackermans, L.; Teernstra, O.P.M.; et al. EGFRvIII expression triggers a metabolic dependency and therapeutic vulnerability sensitive to autophagy inhibition. Autophagy 2018, 14, 283–295.
- Mulcahy Levy, J.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-DeMasters, B.K.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. Elife 2017, 6, e19671.
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.; Fong, H.H.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220.
- Puissant, A.; Robert, G.; Fenouille, N.; Luciano, F.; Cassuto, J.P.; Raynaud, S.; Auberger, P. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010, 70, 1042–1052.
- Selvaraj, S.; Sun, Y.; Sukumaran, P.; Singh, B.B. Resveratrol activates autophagic cell death in prostate cancer cells via downregulation of STIM1 and the mTOR pathway. Mol. Carcinog. 2016, 55, 818–831.
- Zhong, L.X.; Zhang, Y.; Wu, M.L.; Liu, Y.N.; Zhang, P.; Chen, X.Y.; Kong, Q.Y.; Liu, J.; Li, H. Resveratrol and STAT inhibitor enhance autophagy in ovarian cancer cells. Cell Death Discov. 2016, 2, 15071.
- Fukuda, T.; Oda, K.; Wada-Hiraike, O.; Sone, K.; Inaba, K.; Ikeda, Y.; Makii, C.; Miyasaka, A.; Kashiyama, T.; Tanikawa, M.; et al. Autophagy inhibition augments resveratrol-induced apoptosis in Ishikawa endometrial cancer cells. Oncol. Lett. 2016, 12, 2560–2566.
- Dasari, S.K.; Bialik, S.; Levin-Zaidman, S.; Levin-Salomon, V.; Merrill, A.H., Jr.; Futerman, A.H.; Kimchi, A. Signalome-wide RNAi screen identifies GBA1 as a positive mediator of autophagic cell death. Cell Death Differ. 2017, 24, 1288–1302.
- Pyo, J.O.; Jang, M.H.; Kwon, Y.K.; Lee, H.J.; Jun, J.I.; Woo, H.N.; Cho, D.H.; Choi, B.; Lee, H.; Kim, J.H.; et al. Essential roles of Atg5 and FADD in autophagic cell death: Dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 2005, 280, 20722–20729.
- Cea, M.; Cagnetta, A.; Fulciniti, M.; Tai, Y.T.; Hideshima, T.; Chauhan, D.; Roccaro, A.; Sacco, A.; Calimeri, T.; Cottini, F.; et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood 2012, 120, 3519–3529.
- Ginet, V.; Puyal, J.; Rummel, C.; Aubry, D.; Breton, C.; Cloux, A.J.; Majjigapu, S.R.; Sordat, B.; Vogel, P.; Bruzzone, S.; et al. A critical role of autophagy in antileukemia/lymphoma effects of APO866, an inhibitor of NAD biosynthesis. Autophagy 2014, 10, 603–617.
- Lima, S.; Takabe, K.; Newton, J.; Saurabh, K.; Young, M.M.; Leopoldino, A.M.; Hait, N.C.; Roberts, J.L.; Wang, H.G.; Dent, P.; et al. TP53 is required for BECN1- and ATG5-dependent cell death induced by sphingosine kinase 1 inhibition. Autophagy 2018, 14, 942–957.
- Liu, Y.L.; Yang, P.M.; Shun, C.T.; Wu, M.S.; Weng, J.R.; Chen, C.C. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010, 6, 1057–1065.
- Segala, G.; David, M.; de Medina, P.; Poirot, M.C.; Serhan, N.; Vergez, F.; Mougel, A.; Saland, E.; Carayon, K.; Leignadier, J.; et al. Dendrogenin A drives LXR to trigger lethal autophagy in cancers. Nat. Commun. 2017, 8, 1903.
- Kanzawa, T.; Zhang, L.; Xiao, L.; Germano, I.M.; Kondo, Y.; Kondo, S. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene 2005, 24, 980–991.
- Linder, B.; Wehle, A.; Hehlgans, S.; Bonn, F.; Dikic, I.; Rodel, F.; Seifert, V.; Kogel, D. Arsenic Trioxide and (-)-Gossypol Synergistically Target Glioma Stem-Like Cells via Inhibition of Hedgehog and Notch Signaling. Cancers 2019, 11, 350.
- Li, C.L.; Wei, H.L.; Chen, J.; Wang, B.; Xie, B.; Fan, L.L.; Li, L.J. Arsenic trioxide induces autophagy and antitumor effects in Burkitt’s lymphoma Raji cells. Oncol. Rep. 2014, 32, 1557–1563.
- Goussetis, D.J.; Altman, J.K.; Glaser, H.; McNeer, J.L.; Tallman, M.S.; Platanias, L.C. Autophagy is a critical mechanism for the induction of the antileukemic effects of arsenic trioxide. J. Biol. Chem. 2010, 285, 29989–29997.
- Wang, H.; Cao, F.; Li, J.; Li, L.; Li, Y.; Shi, C.; Lan, W.; Li, D.; Zhao, H.; Zhang, Y.; et al. Arsenic trioxide and mannitol for the treatment of acute promyelocytic leukemia relapse in the central nervous system. Blood 2014, 124, 1998–2000.
- Kim, J.; Aftab, B.T.; Tang, J.Y.; Kim, D.; Lee, A.H.; Rezaee, M.; Kim, J.; Chen, B.; King, E.M.; Borodovsky, A.; et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 2013, 23, 23–34.
- Ney, P.A. Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim. Biophys. Acta 2015, 1853(Pt. B), 2775–2783.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.3K
Revisions:
2 times
(View History)
Update Date:
15 Jun 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No