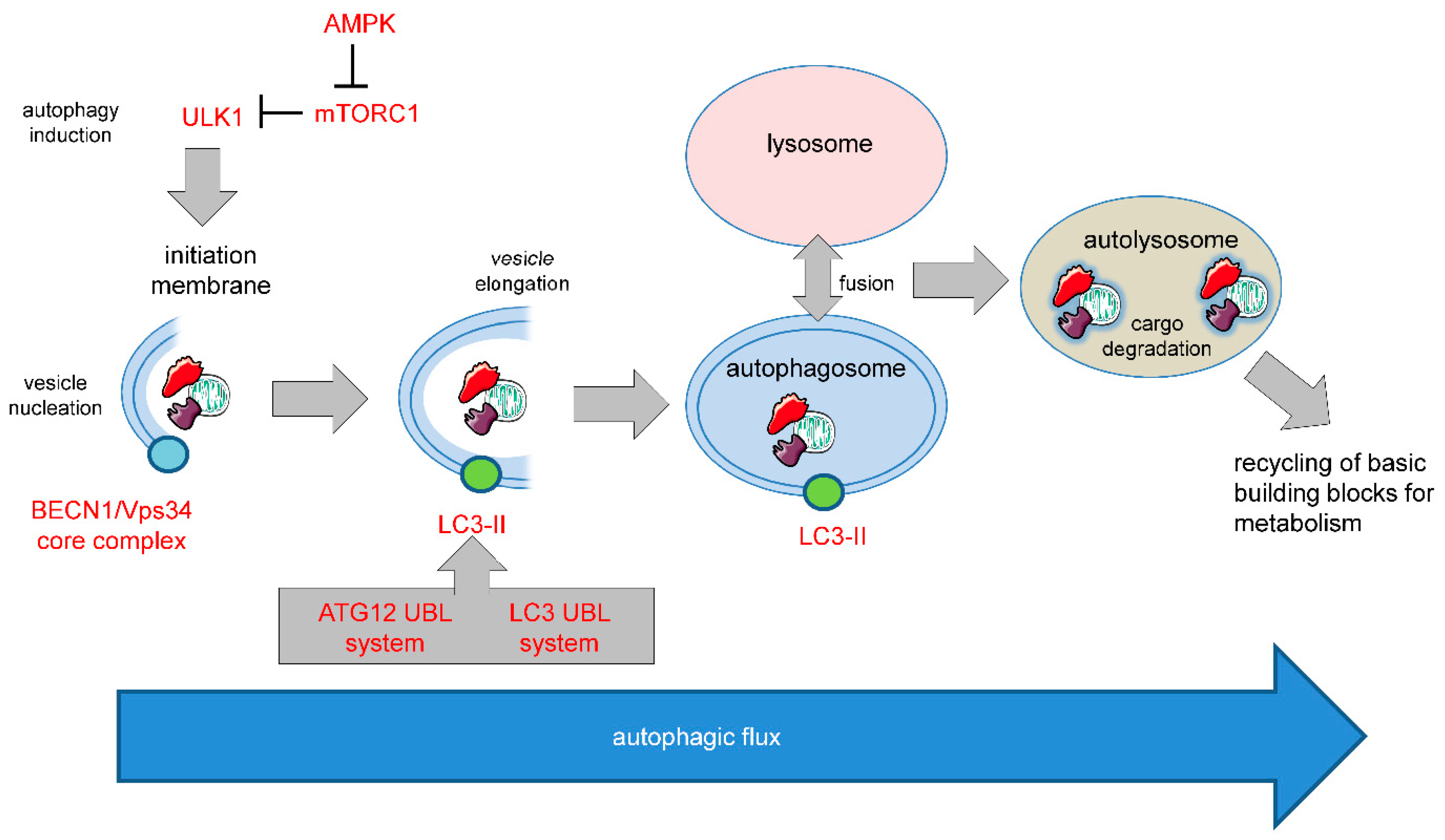
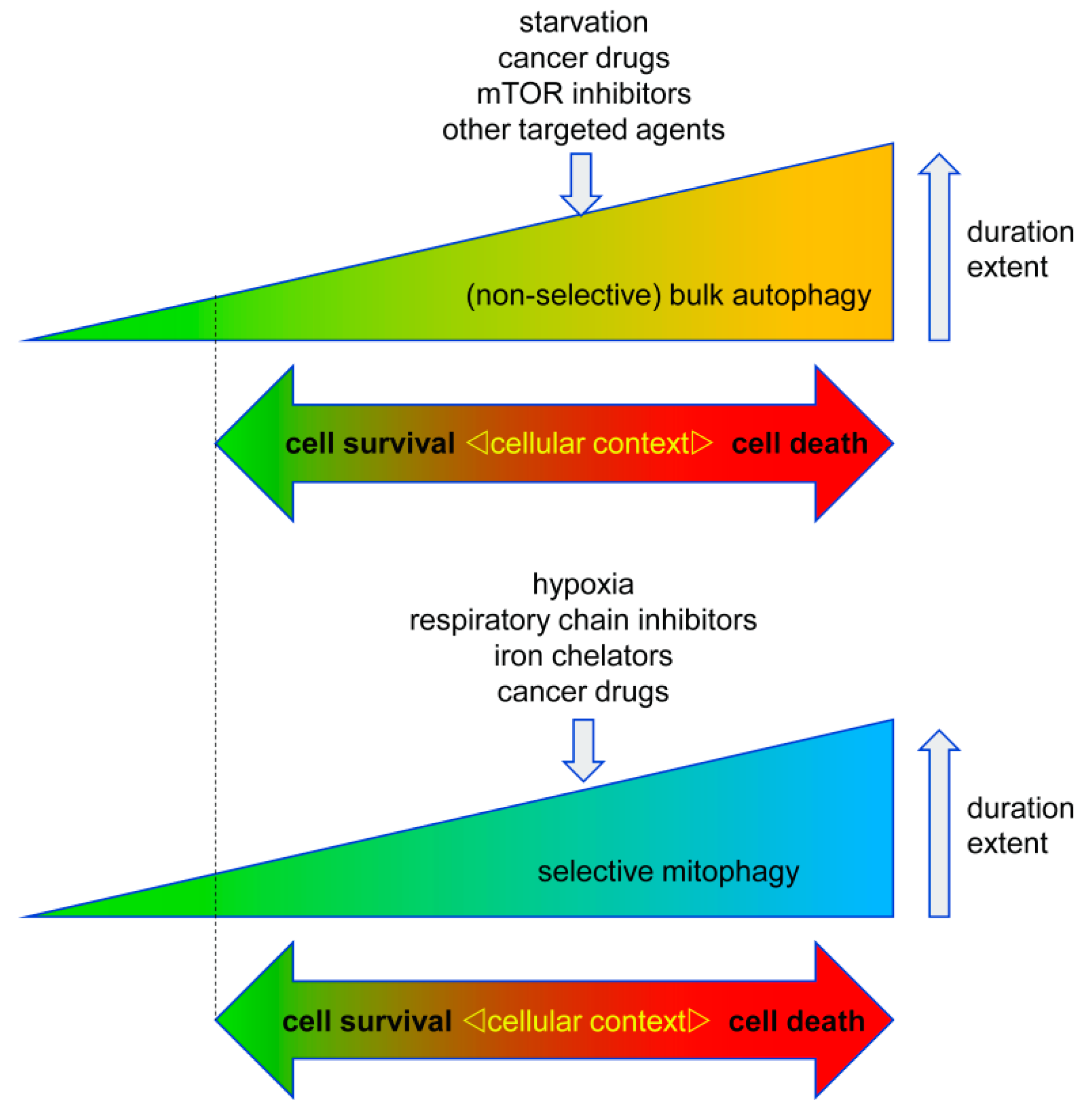
Autophagy has important functions in maintaining energy metabolism under conditions of starvation and to alleviate stress by removal of damaged and potentially harmful cellular components. Therefore, autophagy represents a pro-survival stress response in the majority of cases. An alternative pro-death function of autophagy has been consistently observed in different settings, in particular, in developmental cell death of lower organisms and in drug-induced cancer cell death. This cell death is referred to as autophagic cell death (ACD) or autophagy-dependent cell death (ADCD), a type of cellular demise that may act as a backup cell death program in apoptosis-deficient tumors.
- autophagy
- cell death
- cancer
- gossypol
- AT-101
- mitophagy
- mitochondria
1. Autophagy in Cell Survival and Cell Death
2. Autophagy in Tumorigenesis and Tumor Progression
3. Autophagy in Therapy Response
3.1. Pro-Survival Autophagy
3.2. Pro-Death Autophagy
This entry is adapted from the peer-reviewed paper 10.3390/biology8040082
References
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell. Biol. 2014, 15, 81–94.
- Munoz-Pinedo, C.; Martin, S.J. Autosis: A new addition to the cell death Tower of Babel. Cell Death Dis. 2014, 5, e1319.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541.
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459.
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12 (Suppl. 2), 1509–1518.
- Gozuacik, D.; Kimchi, A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004, 23, 2891–2906.
- Vanzo, R.; Bartkova, J.; Merchut-Maya, J.M.; Hall, A.; Bouchal, J.; Dyrskjot, L.; Frankel, L.B.; Gorgoulis, V.; Maya-Mendoza, A.; Jaattela, M.; et al. Autophagy role(s) in response to oncogenes and DNA replication stress. Cell Death Differ. 2019.
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64.
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410.
- Hamacher-Brady, A.; Brady, N.R.; Gottlieb, R.A. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J. Biol. Chem. 2006, 281, 29776–29787.
- Katheder, N.S.; Khezri, R.; O’Farrell, F.; Schultz, S.W.; Jain, A.; Rahman, M.M.; Schink, K.O.; Theodossiou, T.A.; Johansen, T.; Juhasz, G.; et al. Microenvironmental autophagy promotes tumour growth. Nature 2017, 541, 417–420.
- Monkkonen, T.; Debnath, J. Inflammatory signaling cascades and autophagy in cancer. Autophagy 2018, 14, 190–198.
- Naik, P.P.; Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, C.K.; Mishra, R.; Patil, S.; Bhutia, S.K. Autophagy regulates cisplatin-induced stemness and chemoresistance via the upregulation of CD44, ABCB1 and ADAM17 in oral squamous cell carcinoma. Cell Prolif. 2018, 51, e12411.
- Ran, X.; Zhou, P.; Zhang, K. Autophagy plays an important role in stemness mediation and the novel dual function of EIG121 in both autophagy and stemness regulation of endometrial carcinoma JEC cells. Int. J. Oncol. 2017, 51, 644–656.
- Das, C.K.; Mandal, M.; Kogel, D. Pro-survival autophagy and cancer cell resistance to therapy. Cancer Metastasis Rev. 2018, 37, 749–766.
- Notte, A.; Ninane, N.; Arnould, T.; Michiels, C. Hypoxia counteracts taxol-induced apoptosis in MDA-MB-231 breast cancer cells: Role of autophagy and JNK activation. Cell Death Dis. 2013, 4, e638.
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729.
- Qiu, S.; Sun, L.; Jin, Y.; An, Q.; Weng, C.; Zheng, J. Silencing of BAG3 promotes the sensitivity of ovarian cancer cells to cisplatin via inhibition of autophagy. Oncol. Rep 2017, 38, 309–316.
- DeVorkin, L.; Hattersley, M.; Kim, P.; Ries, J.; Spowart, J.; Anglesio, M.S.; Levi, S.M.; Huntsman, D.G.; Amaravadi, R.K.; Winkler, J.D.; et al. Autophagy Inhibition Enhances Sunitinib Efficacy in Clear Cell Ovarian Carcinoma. Mol. Cancer Res. 2017, 15, 250–258.
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455.
- Pelloski, C.E.; Ballman, K.V.; Furth, A.F.; Zhang, L.; Lin, E.; Sulman, E.P.; Bhat, K.; McDonald, J.M.; Yung, W.K.; Colman, H.; et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J. Clin. Oncol. 2007, 25, 2288–2294.
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009, 16, 748–754.
- Jutten, B.; Keulers, T.G.; Peeters, H.J.M.; Schaaf, M.B.E.; Savelkouls, K.G.M.; Compter, I.; Clarijs, R.; Schijns, O.; Ackermans, L.; Teernstra, O.P.M.; et al. EGFRvIII expression triggers a metabolic dependency and therapeutic vulnerability sensitive to autophagy inhibition. Autophagy 2018, 14, 283–295.
- Mulcahy Levy, J.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-DeMasters, B.K.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. Elife 2017, 6, e19671.
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.; Fong, H.H.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220.
- Puissant, A.; Robert, G.; Fenouille, N.; Luciano, F.; Cassuto, J.P.; Raynaud, S.; Auberger, P. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010, 70, 1042–1052.
- Selvaraj, S.; Sun, Y.; Sukumaran, P.; Singh, B.B. Resveratrol activates autophagic cell death in prostate cancer cells via downregulation of STIM1 and the mTOR pathway. Mol. Carcinog. 2016, 55, 818–831.
- Zhong, L.X.; Zhang, Y.; Wu, M.L.; Liu, Y.N.; Zhang, P.; Chen, X.Y.; Kong, Q.Y.; Liu, J.; Li, H. Resveratrol and STAT inhibitor enhance autophagy in ovarian cancer cells. Cell Death Discov. 2016, 2, 15071.
- Fukuda, T.; Oda, K.; Wada-Hiraike, O.; Sone, K.; Inaba, K.; Ikeda, Y.; Makii, C.; Miyasaka, A.; Kashiyama, T.; Tanikawa, M.; et al. Autophagy inhibition augments resveratrol-induced apoptosis in Ishikawa endometrial cancer cells. Oncol. Lett. 2016, 12, 2560–2566.
- Dasari, S.K.; Bialik, S.; Levin-Zaidman, S.; Levin-Salomon, V.; Merrill, A.H., Jr.; Futerman, A.H.; Kimchi, A. Signalome-wide RNAi screen identifies GBA1 as a positive mediator of autophagic cell death. Cell Death Differ. 2017, 24, 1288–1302.
- Pyo, J.O.; Jang, M.H.; Kwon, Y.K.; Lee, H.J.; Jun, J.I.; Woo, H.N.; Cho, D.H.; Choi, B.; Lee, H.; Kim, J.H.; et al. Essential roles of Atg5 and FADD in autophagic cell death: Dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 2005, 280, 20722–20729.
- Cea, M.; Cagnetta, A.; Fulciniti, M.; Tai, Y.T.; Hideshima, T.; Chauhan, D.; Roccaro, A.; Sacco, A.; Calimeri, T.; Cottini, F.; et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood 2012, 120, 3519–3529.
- Ginet, V.; Puyal, J.; Rummel, C.; Aubry, D.; Breton, C.; Cloux, A.J.; Majjigapu, S.R.; Sordat, B.; Vogel, P.; Bruzzone, S.; et al. A critical role of autophagy in antileukemia/lymphoma effects of APO866, an inhibitor of NAD biosynthesis. Autophagy 2014, 10, 603–617.
- Lima, S.; Takabe, K.; Newton, J.; Saurabh, K.; Young, M.M.; Leopoldino, A.M.; Hait, N.C.; Roberts, J.L.; Wang, H.G.; Dent, P.; et al. TP53 is required for BECN1- and ATG5-dependent cell death induced by sphingosine kinase 1 inhibition. Autophagy 2018, 14, 942–957.
- Liu, Y.L.; Yang, P.M.; Shun, C.T.; Wu, M.S.; Weng, J.R.; Chen, C.C. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010, 6, 1057–1065.
- Segala, G.; David, M.; de Medina, P.; Poirot, M.C.; Serhan, N.; Vergez, F.; Mougel, A.; Saland, E.; Carayon, K.; Leignadier, J.; et al. Dendrogenin A drives LXR to trigger lethal autophagy in cancers. Nat. Commun. 2017, 8, 1903.
- Kanzawa, T.; Zhang, L.; Xiao, L.; Germano, I.M.; Kondo, Y.; Kondo, S. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene 2005, 24, 980–991.
- Linder, B.; Wehle, A.; Hehlgans, S.; Bonn, F.; Dikic, I.; Rodel, F.; Seifert, V.; Kogel, D. Arsenic Trioxide and (-)-Gossypol Synergistically Target Glioma Stem-Like Cells via Inhibition of Hedgehog and Notch Signaling. Cancers 2019, 11, 350.
- Li, C.L.; Wei, H.L.; Chen, J.; Wang, B.; Xie, B.; Fan, L.L.; Li, L.J. Arsenic trioxide induces autophagy and antitumor effects in Burkitt’s lymphoma Raji cells. Oncol. Rep. 2014, 32, 1557–1563.
- Goussetis, D.J.; Altman, J.K.; Glaser, H.; McNeer, J.L.; Tallman, M.S.; Platanias, L.C. Autophagy is a critical mechanism for the induction of the antileukemic effects of arsenic trioxide. J. Biol. Chem. 2010, 285, 29989–29997.
- Wang, H.; Cao, F.; Li, J.; Li, L.; Li, Y.; Shi, C.; Lan, W.; Li, D.; Zhao, H.; Zhang, Y.; et al. Arsenic trioxide and mannitol for the treatment of acute promyelocytic leukemia relapse in the central nervous system. Blood 2014, 124, 1998–2000.
- Kim, J.; Aftab, B.T.; Tang, J.Y.; Kim, D.; Lee, A.H.; Rezaee, M.; Kim, J.; Chen, B.; King, E.M.; Borodovsky, A.; et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 2013, 23, 23–34.
- Ney, P.A. Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim. Biophys. Acta 2015, 1853(Pt. B), 2775–2783.