 +1 credit
+1 credit
Video Upload Options
Sickle cell disease (SCD) is the most common hereditary disorder of hemoglobin (Hb), which affects approximately a million people worldwide. It is characterized by a single nucleotide substitution in the β-globin gene, leading to the production of abnormal sickle hemoglobin (HbS) with multi-system consequences. HbS polymerization is the primary event in SCD. Repeated polymerization and depolymerization of Hb causes oxidative stress that plays a key role in the pathophysiology of hemolysis, vessel occlusion and the following organ damage in sickle cell patients. For this reason, reactive oxidizing species and the (end)-products of their oxidative reactions have been proposed as markers of both tissue pro-oxidant status and disease severity. Although more studies are needed to clarify their role, antioxidant agents have been shown to be effective in reducing pathological consequences of the disease by preventing oxidative damage in SCD, i.e., by decreasing the oxidant formation or repairing the induced damage.
1. Introduction
2. Source of ROS in SCD
2.1. Increased Activity of Several Oxidases
2.2. HbS Autoxidation
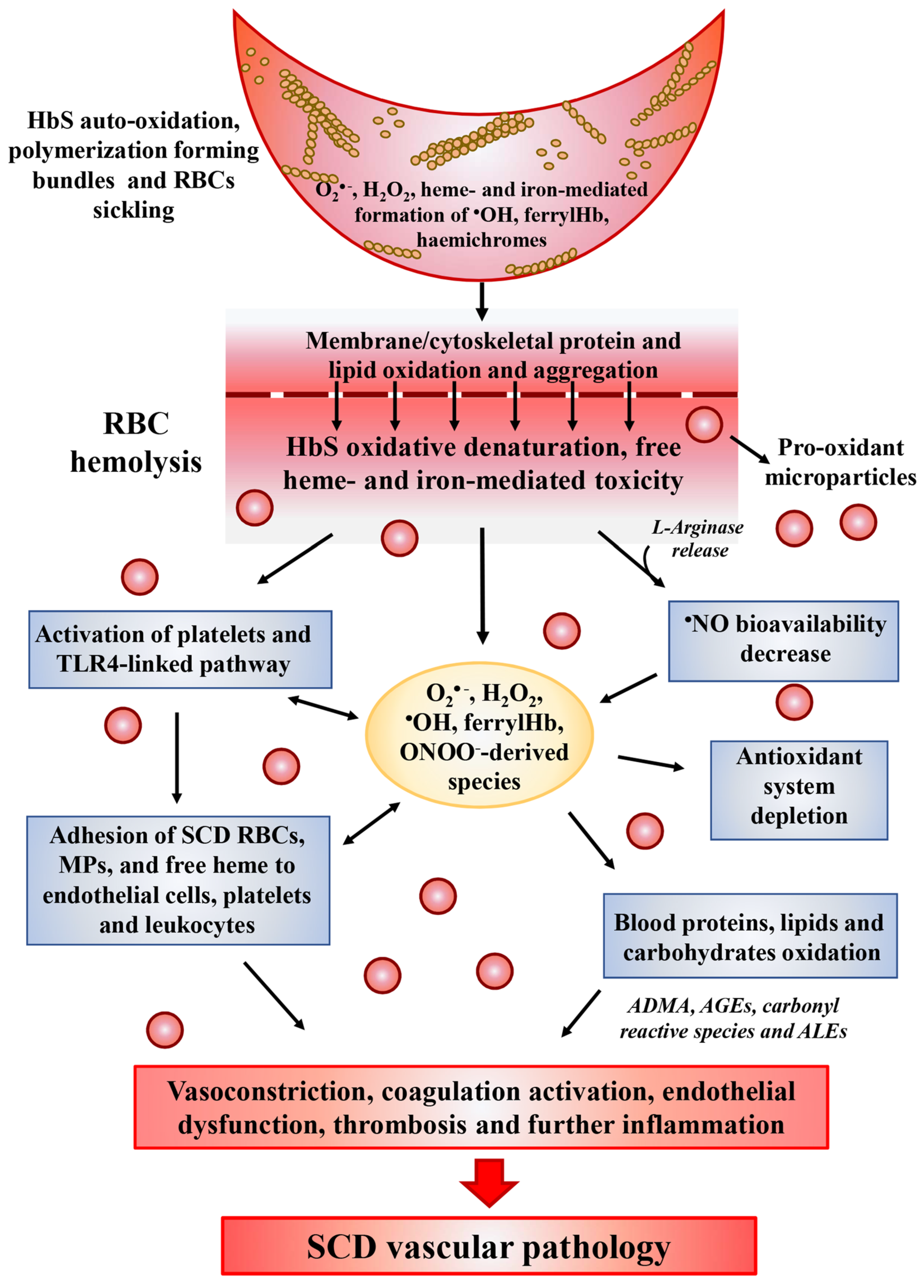
2.3. Heme and Iron Release
2.4. Decreased •NO Bioavailability
3. Antioxidant Therapy for SCD
3.1. L-Glutamine
| Antioxidants | Mechanisms | Effects in SCD Patients | Comments |
|---|---|---|---|
| L- Glutamine | Acts through the formation of reduced NADH. | Improves cellular redox potential and adhesion of sickle RBCs to the endothelium; facilitates protein and glutathione synthesis. | FDA approved; [52][57] |
| N-Acetylcysteine | Substrate for GSH generation. | Reduces the number of RBCs expressing phosphatidylserine, marker of peroxidative damage to inner membrane of RBCs. | NCT01800526 phase 2 trial; NCT01849016 phase 3 trial |
| Zinc supplementation | Zinc deficiency is associated with high incidence of infections, vaso-occlusion events and the chronic oxidative stress. | In young SCD patients improves linear growth and weight gain. Has beneficial effects on immunity, inflammatory state and oxidative stress. | NS; [56][58] |
| Nitric oxide | Reduced •NO concentration can be associated to increased levels of free O2•−. | Inhaled •NO improves tissue oxygenation and reduces pain in SCD patients with pulmonary hypertension. | NCT00094887 phase 2 trial |
| L-arginine | Induces GSH synthesis. | Improves •NO bioavailability. | NCT02447874 phase 2 trial |
| Alfa-lipoic acid | Induces GSH synthesis. | Increases glutathione level. | NCT01054768 phase 2 trial |
| L-acetyl-L-carnitine | Improves mitochondrial metabolism, facilitating entry of long-chain fatty acids into mitochondria and decreasing lipid peroxidation in tissue. | Decreases lipid peroxidation. | NCT01054768 phase 2 trial |
| Gum Arabic | Acts as immuno-modulatory. | Increases total antioxidant capacity and decreased MDA and H2O2 levels. | NCT04191213 phase 2 trial |
| Omega-3 fatty acids | O3FA deficiency correlates with an increase in plasma levels of the inflammatory biomarker | Have beneficial effects on vascular activation, inflammation and antioxidant systems. | [52][53][55][56][58][59][60][61][62][63][64][65][66][67][68] |
| Curcumin | Can modulate the activity of enzymes active in the neutralization of free radicals and it can inhibit ROS-generating enzymes. | Mitigates the effects of iron induced oxidative stress on lipid peroxidation and •NO levels. | [67][68] |
| Vitamins A, C and E | Their deficiency increases susceptibility to infection and hemolysis. | Conflicting results about the effectiveness of their supplementation on oxidative stress. | NCT03903133 phase 4 trial |
| Iron chelators | Avoids excessive iron overload and the consequent ROS generation. | Have a central role in the treatment of transfusion-dependent hemoglobinopathy. | NS; [55] |
3.2. N-Acetylcysteine (NAC)
3.3. Zinc Supplementation
3.4. Nitric Oxide and L-Arginine
3.5. α-Lipoic Acid and Acetyl-L-Carnitine
3.6. Other Antioxidant Agents
References
- Chaturvedi, S.; DeBaun, M.R. Evolution of sickle cell disease from a life-threatening disease of children to a chronic disease of adults: The last 40 years. Am. J. Hematol. 2016, 91, 5–14.
- Thein, M.S.; Igbineweka, N.E.; Thein, S.L. Sickle cell disease in the older adult. Pathology 2017, 49, 1–9.
- Aygun, B.; Odame, I. A global perspective on sickle cell disease. Pediatr. Blood Cancer 2012, 59, 386–390.
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat. Rev. Dis. Primers 2018, 4, 18010.
- Piel, F.B.; Steinberg, M.H.; Rees, D.C. Sickle Cell Disease. N. Engl. J. Med. 2017, 377, 305.
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031.
- Ashley-Koch, A.; Yang, Q.; Olney, R.S. Sickle hemoglobin (HbS) allele and sickle cell disease: A HuGE review. Am. J. Epidemiol. 2000, 151, 839–845.
- Stuart, M.J.; Nagel, R.L. Sickle-cell disease. Lancet 2004, 364, 1343–1360.
- Tarasev, M.; Muchnik, M.; Light, L.; Alfano, K.; Chakraborty, S. Individual variability in response to a single sickling event for normal, sickle cell, and sickle trait erythrocytes. Transl. Res. 2017, 181, 96–107.
- Chirico, E.N.; Pialoux, V. Role of oxidative stress in the pathogenesis of sickle cell disease. IUBMB Life 2012, 64, 72–80.
- Dosunmu-Ogunbi, A.M.; Wood, K.C.; Novelli, E.M.; Straub, A.C. Decoding the role of SOD2 in sickle cell disease. Blood Adv. 2019, 3, 2679–2687.
- Steinberg, M.H.; Sebastiani, P. Genetic modifiers of sickle cell disease. Am. J. Hematol. 2012, 87, 795–803.
- Ware, R.E.; de Montalembert, M.; Tshilolo, L.; Abboud, M.R. Sickle cell disease. Lancet 2017, 390, 311–323.
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462.
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183.
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848.
- Ray, D.; Deshmukh, P.; Goswami, K.; Garg, N. Antioxidant vitamin levels in sickle cell disorders. Natl. Med. J. India 2007, 20, 11–13.
- Biswal, S.; Rizwan, H.; Pal, S.; Sabnam, S.; Parida, P.; Pal, A. Oxidative stress, antioxidant capacity, biomolecule damage, and inflammation symptoms of sickle cell disease in children. Hematology 2019, 24, 1–9.
- Al-Naama, L.M.; Hassan, M.K.; Mehdi, J.K. Association of erythrocytes antioxidant enzymes and their cofactors with markers of oxidative stress in patients with sickle cell anemia. Qatar Med. J. 2015, 2015, 14.
- Cardenes, N.; Corey, C.; Geary, L.; Jain, S.; Zharikov, S.; Barge, S.; Novelli, E.M.; Shiva, S. Platelet bioenergetic screen in sickle cell patients reveals mitochondrial complex V inhibition, which contributes to platelet activation. Blood 2014, 123, 2864–2872.
- Wood, K.C.; Hebbel, R.P.; Granger, D.N. Endothelial cell NADPH oxidase mediates the cerebral microvascular dysfunction in sickle cell transgenic mice. FASEB J. 2005, 19, 989–991.
- Wood, K.C.; Hebbel, R.P.; Lefer, D.J.; Granger, D.N. Critical role of endothelial cell-derived nitric oxide synthase in sickle cell disease-induced microvascular dysfunction. Free Radic. Biol. Med. 2006, 40, 1443–1453.
- Wood, K.C.; Granger, D.N. Sickle cell disease: Role of reactive oxygen and nitrogen metabolites. Clin. Exp. Pharmacol. Physiol. 2007, 34, 926–932.
- George, A.; Pushkaran, S.; Konstantinidis, D.G.; Koochaki, S.; Malik, P.; Mohandas, N.; Zheng, Y.; Joiner, C.H.; Kalfa, T.A. Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood 2013, 121, 2099–2107.
- Aslan, M.; Ryan, T.M.; Adler, B.; Townes, T.M.; Parks, D.A.; Thompson, J.A.; Tousson, A.; Gladwin, M.T.; Patel, R.P.; Tarpey, M.M.; et al. Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proc. Natl. Acad. Sci. USA 2001, 98, 15215–15220.
- Schaer, D.J.; Buehler, P.W.; Alayash, A.I.; Belcher, J.D.; Vercellotti, G.M. Hemolysis and free hemoglobin revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 2013, 121, 1276–1284.
- Rifkind, J.M.; Mohanty, J.G.; Nagababu, E. The pathophysiology of extracellular hemoglobin associated with enhanced oxidative reactions. Front. Physiol. 2014, 5, 500.
- Gizi, A.; Papassotiriou, I.; Apostolakou, F.; Lazaropoulou, C.; Papastamataki, M.; Kanavaki, I.; Kalotychou, V.; Goussetis, E.; Kattamis, A.; Rombos, I.; et al. Assessment of oxidative stress in patients with sickle cell disease: The glutathione system and the oxidant-antioxidant status. Blood Cells Mol. Dis. 2011, 46, 220–225.
- Kassa, T.; Jana, S.; Strader, M.B.; Meng, F.; Jia, Y.; Wilson, M.T.; Alayash, A.I. Sickle Cell Hemoglobin in the Ferryl State Promotes betaCys-93 Oxidation and Mitochondrial Dysfunction in Epithelial Lung Cells (E10). J. Biol. Chem. 2015, 290, 27939–27958.
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O., 3rd; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389.
- Berlett, B.S.; Stadtman, E.R. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 1997, 272, 20313–20316.
- Mano, J. Reactive carbonyl species: Their production from lipid peroxides, action in environmental stress, and the detoxification mechanism. Plant. Physiol. Biochem. 2012, 59, 90–97.
- Azarov, I.; He, X.; Jeffers, A.; Basu, S.; Ucer, B.; Hantgan, R.R.; Levy, A.; Kim-Shapiro, D.B. Rate of nitric oxide scavenging by hemoglobin bound to haptoglobin. Nitric Oxide 2008, 18, 296–302.
- Nielsen, M.J.; Moller, H.J.; Moestrup, S.K. Hemoglobin and heme scavenger receptors. Antioxid. Redox Signal. 2010, 12, 261–273.
- Koskenkorva-Frank, T.S.; Weiss, G.; Koppenol, W.H.; Burckhardt, S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: Insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic. Biol. Med. 2013, 65, 1174–1194.
- Balla, J.; Jacob, H.S.; Balla, G.; Nath, K.; Eaton, J.W.; Vercellotti, G.M. Endothelial-cell heme uptake from heme proteins: Induction of sensitization and desensitization to oxidant damage. Proc. Natl. Acad. Sci. USA 1993, 90, 9285–9289.
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188.
- Belcher, J.D.; Beckman, J.D.; Balla, G.; Balla, J.; Vercellotti, G. Heme degradation and vascular injury. Antioxid. Redox Signal. 2010, 12, 233–248.
- Aslan, M.; Freeman, B.A. Oxidant-mediated impairment of nitric oxide signaling in sickle cell disease—Mechanisms and consequences. Cell Mol. Biol. 2004, 50, 95–105.
- Jeffers, A.; Gladwin, M.T.; Kim-Shapiro, D.B. Computation of plasma hemoglobin nitric oxide scavenging in hemolytic anemias. Free Radic. Biol. Med. 2006, 41, 1557–1565.
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Investig. 2017, 127, 750–760.
- Shiva, S.; Wang, X.; Ringwood, L.A.; Xu, X.; Yuditskaya, S.; Annavajjhala, V.; Miyajima, H.; Hogg, N.; Harris, Z.L.; Gladwin, M.T. Ceruloplasmin is a NO oxidase and nitrite synthase that determines endocrine NO homeostasis. Nat. Chem. Biol. 2006, 2, 486–493.
- Grubina, R.; Basu, S.; Tiso, M.; Kim-Shapiro, D.B.; Gladwin, M.T. Nitrite reductase activity of hemoglobin S (sickle) provides insight into contributions of heme redox potential versus ligand affinity. J. Biol. Chem. 2008, 283, 3628–3638.
- Rees, D.C.; Gibson, J.S. Biomarkers in sickle cell disease. Br. J. Haematol. 2012, 156, 433–445.
- Antwi-Boasiako, C.; Dankwah, G.B.; Aryee, R.; Hayfron-Benjamin, C.; Aboagye, G.; Campbell, A.D. Correlation of lipid peroxidation and nitric oxide metabolites, trace elements, and antioxidant enzymes in patients with sickle cell disease. J. Clin. Lab. Anal. 2020, 34, e23294.
- Reiter, C.D.; Gladwin, M.T. An emerging role for nitric oxide in sickle cell disease vascular homeostasis and therapy. Curr. Opin. Hematol. 2003, 10, 99–107.
- Azizi, E.; Dror, Y.; Wallis, K. Arginase activity in erythrocytes of healthy and ill children. Clin. Chim Acta 1970, 28, 391–396.
- Morris, C.R.; Kato, G.J.; Poljakovic, M.; Wang, X.; Blackwelder, W.C.; Sachdev, V.; Hazen, S.L.; Vichinsky, E.P.; Morris, S.M., Jr.; Gladwin, M.T. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA 2005, 294, 81–90.
- Elias, D.B.; Barbosa, M.C.; Rocha, L.B.; Dutra, L.L.; Silva, H.F.; Martins, A.M.; Goncalves, R.P. L-arginine as an adjuvant drug in the treatment of sickle cell anaemia. Br. J. Haematol. 2013, 160, 410–412.
- Davies, M.J.; Hawkins, C.L. The Role of Myeloperoxidase in Biomolecule Modification, Chronic Inflammation, and Disease. Antioxid. Redox Signal. 2020, 32, 957–981.
- Baldus, S.; Eiserich, J.P.; Brennan, M.L.; Jackson, R.M.; Alexander, C.B.; Freeman, B.A. Spatial mapping of pulmonary and vascular nitrotyrosine reveals the pivotal role of myeloperoxidase as a catalyst for tyrosine nitration in inflammatory diseases. Free Radic. Biol. Med. 2002, 33, 1010.
- Kapoor, S.; Little, J.A.; Pecker, L.H. Advances in the Treatment of Sickle Cell Disease. Mayo Clin. Proc. 2018, 93, 1810–1824.
- Niihara, Y.; Macan, H.; Eckman, J.R.; Koh, H.; Cooper, M.L.; Ziegler, T.R.; Razon, R.; Tanaka, K.R.; Stark, C.W.; Johnson, C.S. L-Glutamine Therapy Reduces Hospitalization for Sickle Cell Anemia and Sickle β°-Thalassemia Patients at Six Months—A Phase II Randomized Trial. Clin. Pharmacol. Biopharm. 2014, 3, 116.
- Zerez, C.R.; Lachant, N.A.; Lee, S.J.; Tanaka, K.R. Decreased erythrocyte nicotinamide adenine dinucleotide redox potential and abnormal pyridine nucleotide content in sickle cell disease. Blood 1988, 71, 512–515.
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N. Engl. J. Med. 2018, 379, 226–235.
- Ortiz de Montellano, P.R. A New Step in the Treatment of Sickle Cell DiseasePublished as part of the Biochemistry series “Biochemistry to Bedside”. Biochemistry 2018, 57, 470–471.
- Vendrame, F.; Olops, L.; Saad, S.T.O.; Costa, F.F.; Fertrin, K.Y. Differences in heme and hemopexin content in lipoproteins from patients with sickle cell disease. J. Clin. Lipidol. 2018, 12, 1532–1538.
- Silva, D.G.H.; Belini Junior, E.; de Almeida, E.A.; Bonini-Domingos, C.R. Oxidative stress in sickle cell disease: An overview of erythrocyte redox metabolism and current antioxidant therapeutic strategies. Free Radic. Biol. Med. 2013, 65, 1101–1109.
- Zemel, B.S.; Kawchak, D.A.; Fung, E.B.; Ohene-Frempong, K.; Stallings, V.A. Effect of zinc supplementation on growth and body composition in children with sickle cell disease. Am. J. Clin. Nutr. 2002, 75, 300–307.
- Bao, B.; Prasad, A.S.; Beck, F.W.; Snell, D.; Suneja, A.; Sarkar, F.H.; Doshi, N.; Fitzgerald, J.T.; Swerdlow, P. Zinc supplementation decreases oxidative stress, incidence of infection, and generation of inflammatory cytokines in sickle cell disease patients. Transl. Res. 2008, 152, 67–80.
- Aboursheid, T.; Albaroudi, O.; Alahdab, F. Inhaled nitric oxide for treating pain crises in people with sickle cell disease. Cochrane Database Syst. Rev. 2019, 10, CD011808.
- Dasgupta, T.; Hebbel, R.P.; Kaul, D.K. Protective effect of arginine on oxidative stress in transgenic sickle mouse models. Free Radic. Biol. Med. 2006, 41, 1771–1780.
- Kaul, D.K.; Zhang, X.; Dasgupta, T.; Fabry, M.E. Arginine therapy of transgenic-knockout sickle mice improves microvascular function by reducing non-nitric oxide vasodilators, hemolysis, and oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H39–H47.
- Vasquez-Vivar, J. Tetrahydrobiopterin, superoxide, and vascular dysfunction. Free Radic. Biol. Med. 2009, 47, 1108–1119.
- Lal, A.; Suh, J.H.; Atamna, W.; Canty, B.; Hagar, W.; Vichinsky, E.F.; Ames, B. Anti-oxidant treatment with alipoic acid and acetyl L-carnitine in hemoglobinopathies. Blood 2007, 11, 3799.
- Lal, A.; Atamna, W.; Killilea, D.W.; Suh, J.H.; Ames, B.N. Lipoic acid and acetyl-carnitine reverse iron-induced oxidative stress in human fibroblasts. Redox Rep. 2008, 13, 2–10.
- Kaddam, L.; Fadl-Elmula, I.; Eisawi, O.A.; Abdelrazig, H.A.; Salih, M.A.; Lang, F.; Saeed, A.M. Gum Arabic as novel anti-oxidant agent in sickle cell anemia, phase II trial. BMC Hematol. 2017, 17, 4.
- Kalish, B.T.; Matte, A.; Andolfo, I.; Iolascon, A.; Weinberg, O.; Ghigo, A.; Cimino, J.; Siciliano, A.; Hirsch, E.; Federti, E.; et al. Dietary omega-3 fatty acids protect against vasculopathy in a transgenic mouse model of sickle cell disease. Haematologica 2015, 100, 870–880.
- Nur, E.; Brandjes, D.P.; Teerlink, T.; Otten, H.M.; Oude Elferink, R.P.; Muskiet, F.; Evers, L.M.; Ten Cate, H.; Biemond, B.J.; Duits, A.J.; et al. N-acetylcysteine reduces oxidative stress in sickle cell patients. Ann. Hematol. 2012, 91, 1097–1105.
- Potoka, K.P.; Gladwin, M.T. Vasculopathy and pulmonary hypertension in sickle cell disease. Am. J. Physiol. Lung Cell. Mol. Physiol 2015, 308, L314–L324.
- Badria, F.A.; Ibrahim, A.S.; Badria, A.F.; Elmarakby, A.A. Curcumin Attenuates Iron Accumulation and Oxidative Stress in the Liver and Spleen of Chronic Iron-Overloaded Rats. PLoS ONE 2015, 10, e0134156.
- Badria, F.A.; Ibrahim, A.S.; Badria, A.F.; Elmarakby, A.A. Correction: Curcumin attenuates iron accumulation and oxidative stress in the liver and spleen of chronic iron-overloaded rats. PLoS ONE 2020, 15, e0243398.
- Belini Junior, E.; da Silva, D.G.; Torres Lde, S.; de Almeida, E.A.; Cancado, R.D.; Chiattone, C.; Bonini-Domingos, C.R. Oxidative stress and antioxidant capacity in sickle cell anaemia patients receiving different treatments and medications for different periods of time. Ann. Hematol. 2012, 91, 479–489.


