Sickle cell disease (SCD) is the most common hereditary disorder of hemoglobin (Hb), which affects approximately a million people worldwide. It is characterized by a single nucleotide substitution in the β-globin gene, leading to the production of abnormal sickle hemoglobin (HbS) with multi-system consequences. HbS polymerization is the primary event in SCD. Repeated polymerization and depolymerization of Hb causes oxidative stress that plays a key role in the pathophysiology of hemolysis, vessel occlusion and the following organ damage in sickle cell patients. For this reason, reactive oxidizing species and the (end)-products of their oxidative reactions have been proposed as markers of both tissue pro-oxidant status and disease severity. Although more studies are needed to clarify their role, antioxidant agents have been shown to be effective in reducing pathological consequences of the disease by preventing oxidative damage in SCD, i.e., by decreasing the oxidant formation or repairing the induced damage.
- sickle cell disease
- hemoglobin
- oxidative stress
- antioxidants
- red blood cells
1. Introduction
2. Source of ROS in SCD
In SCD, reactive oxidizing species are generated by sickle RBCs as well as by activated leukocytes, platelets, ECs, and plasma enzymes. Several mechanisms contribute to ROS and RNS formation in tissues of SCD patients such as (i) increased activity of nicotinamide-adenine dinucleotide phosphate (NADPH) oxidase and endothelial xanthine oxidase (XO) [8[8][10],10], (ii) HbS autoxidation [11], (iii) heme and iron release, (iv) increased asymmetric dimethylarginine (ADMA) [12[12][13],13], and (v) uncoupling of nitric oxide synthase (NOS) activity and decreased •NO bioavailability [20].2.1. Increased Activity of Several Oxidases
It has been demonstrated that in SCD, the enzymes NADPH oxidase, XO and uncoupled endothelial nitric oxide synthase (eNOS) can generate ROS in the vascular compartment [21,22,23,24][21][22][23][24]. NADPH oxidase is the major O2•− producing enzyme in leucocytes, vascular endothelial cells and RBCs. ROS produced by activated leucocyte NADPH oxidase contribute to the hemolysis associated with infections or vessel occlusive crises [23]. The O2•−, derived from endothelial cell NADPH oxidase, contributes to the pro-inflammatory and pro-thrombogenic responses associated with SCD [24]. In RBCs, NADPH oxidase activity is regulated intracellularly by protein kinase C and Rac GTPases and extracellularly by signaling factors such as transforming growth factor β1 and endothelin-1 present in the plasma from SCD patients [24]. ROS derived by RBC NADPH oxidase may cause direct oxidative damage to a variety of subcellular structures, reducing RBC deformability and resulting in increased RBC fragility and hemolysis [24]. XO represents a potent source of superoxide O2•− and H2O2, and its activity is increased in the plasma of SCD patients. The source of XO is not completely clear, but episodes of hypoxia/reoxygenation in SCD patients can stimulate the release of this enzyme from the liver into the circulation. Increased circulating XO can then bind avidly to vessel luminal cells and impairing vascular function and creating an oxidative milieu [25].2.2. HbS Autoxidation
Normal RBCs continuously generate ROS during oxygenation/deoxygenation cycles occurring in the circulation. The oxygen exchange physiologically generates a continuous slow autoxidation of oxygenated Hb (ferrous, Hb-FeII) producing O2•− and methemoglobin (ferric, Hb-FeIII), which no longer binds oxygen, at a rate of 0.5–3% per day. The spontaneous and enzymatic O2•− dismutation forms H2O2, but both these species are neutralized by the efficient RBC antioxidant system involving both non-enzymatic low molecular weight antioxidants (glutathione, ascorbic acid and vitamin E) and enzymatic antioxidants (SOD, Cat, GR, Prx2 and Gpx). These antioxidant activities, coupled to the methemoglobin reductase-dependent reduction of Hb-FeIII to Hb-FeII, preserve RBCs integrity and function. Under conditions of oxidative stress, ROS are produced in greater quantities in normal RBCs, which by activating the pseudo-peroxidase cycle detoxify the generated oxidants leading to the complete consumption of H2O2. In SCD, intravascular hemolysis results in the toxic accumulation of free HbS and heme in the plasma (Figure 1). Compared to normal Hb, HbS molecules are highly unstable in particular under hypoxic condition and more prone to autoxidation [26,27][26][27]. The rate of HbS autoxidation has been calculated to be about 2 times faster than that of normal Hb, resulting in the increase of about 2 times the generation of O2•−, H2O2, •OH and lipid oxidation products [28,29][28][29]. This exacerbated pseudo-peroxidase cycle is followed by heme release and iron loss, both able to amplify oxidative reactions. In addition, the autoreduction of ferryl back to ferric heme is slower than that of normal Hb, leading to a longer lived and more damaging free ferryl Hb and to free ferryl radical. The latter has been shown to migrate and induce further damage in the protein, including the irreversible oxidation and dimerization of Cysβ93, as well as to induce, in target cells, damage and dysfunction in other biological organelles, such as in the mitochondria likely, contributing to SCD-induced vascular pathology [29]. In SCD, as well as in other hemolityc disorders, the high amounts of plasma hemoglobin is in the ferrous form and can stoichiometrically react with equivalent amounts of •NO [30]. This reaction leads to the in vivo formation of •NO–hemoglobin deoxygenation end-products (methemoglobin, nitrate) and iron–nitrosylhemoglobin complexes, thus contributing to decrease •NO bioavailability [30].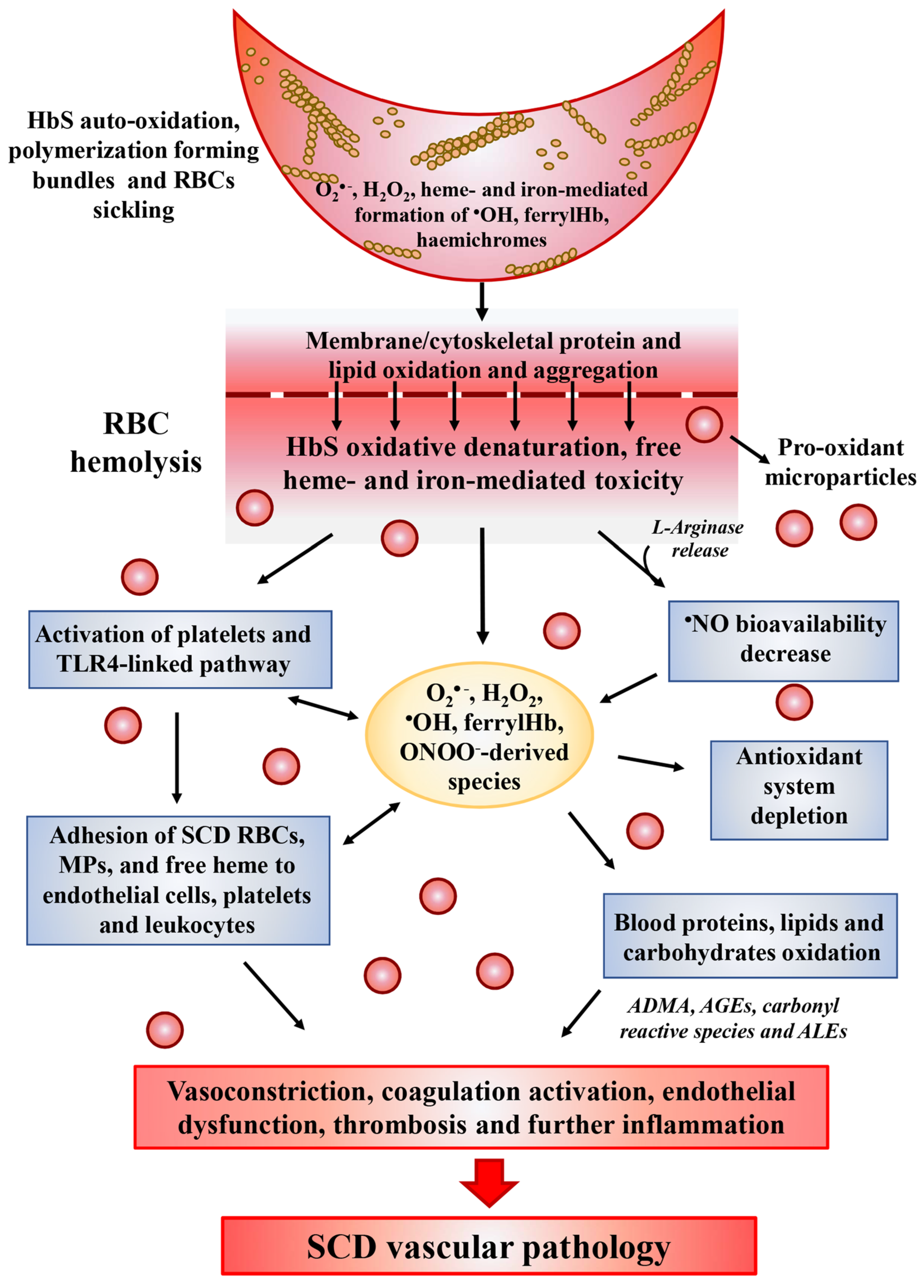
2.3. Heme and Iron Release
Under mild to moderate hemolysis, Hb is bound in plasma by haptoglobin (Hp) forming a complex, which prevents the release of free iron while continuing to maintain the ability to bind •NO [31][33]. The complex is internalized and degraded through the CD163 receptor found on macrophages and CD91 receptors found on hepatocytes [32][34]. The release of heme from HbS is faster than from normal Hb [29]. Its characteristic hydrophobicity allows heme to intercalate into the cell membranes and magnify the intracellular heme-dependent reactive oxidizing species generation. In addition, under inflammatory condition, O2•− and H2O2, released by activated cells, can react with heme and catalyze both the non-enzymatic generation of reactive oxidizing species as well as the release of free reducing ferrous ions, which in turn may increase the Fenton-driven reactions and induce further oxidative and nitrosative stress [33][35] (Figure 1). These events amplify the formation of ROS inside cells leading to additional damage to intracellular components including proteins, lipids, and DNA [34,35][36][37]. Consequently, fundamental functions of cells may be compromised by this heme- and iron-mediated increase of ROS formation, such as the intracellular signaling mediated by oxidant-sensitive targets, the expression of pro-inflammatory transcription factors, the integrity of membrane channels, the activity of metabolic enzymes, inducing finally cell apoptosis and death. In addition, heme-derived oxidants induce recruitment of leukocytes, platelets and RBCs to the vessel wall; produce lipoproteins oxidation and consume •NO in the formation of strong oxidants such as ONOO− (Figure 1) [36][38].2.4. Decreased
•
NO Bioavailability
In SCD, all •NO biological functions, including the regulation of vascular tone, the control of cell activation, aggregation and adhesion in the vascular compartment, are compromised so that vasoconstriction, pulmonary hypertension, endothelial dysfunction, thrombosis and inflammation characterize the vasculopathy linked to this disease [39,40,41][39][40][41]. In the absence of interactions with biological targets, •NO reacts with oxygen (9 × 107 M−1s−1) to form nitrite. In biological systems, this reaction is slower with respect to those occurring with metal-containing macromolecules generating either nitrate, as in the case of hemoglobin (6–8 × 107 M−1s−1) and nitrite in the case of ceruloplasmin acting as a •NO oxidase and nitrite synthase [42]. Nitrite is not an inert end-product, but it has bioactivity since it can generate •NO from its reaction with metalloproteins, as in the case of HbS itself, which has been reported to possess nitrite reductase activity [43]. In addition, the concentration of these •NO-derived metabolites is deeply affected by the dietary intake and renal function. In plasma of SCD patients, the concentrations of nitrite and nitrate appear to undergo modifications, i.e., in the steady state of disease they have been found to be comparable to those measured in normal volunteers, but they decreased with acute pain and acute chest syndrome [44,45][44][45]. The impairment of •NO availability in SCD is mainly linked to the intravascular hemolysis (Figure 1). Cell-free hemoglobin has indeed a large impact on the bioavailability of •NO. While the reaction of •NO with oxygenated Hb results in methemoglobin and nitrate formation, its binding to deoxygenated hemoglobin favors the formation of a stable FeIIHb-NO complex, which can be involved in Fenton reactions [36][38]. Interestingly, a gender difference was also described in •NO availability in SCD patients. In particular, thanks to the protective effects of estrogen on the expression and activity of NOS, women are more protected from the decrease of •NO availability [46]. Another crucial metabolite contributing to vascular impairment in SCD is L-arginine. Besides being used by NOS to generate •NO, L-arginine is also the substrate of the enzyme arginase, which competes with NOS for L-arginine, generating urea and ornithine. Since the arginase activity is significantly high in RBCs compared to plasma [47], the hemolysis causes the release of the enzyme from RBCs into plasma allowing to the rapid consumption of L-arginine, reducing the substrate for •NO synthesis and consequently its bioavailability in SCD (Figure 1) [48]. In SCD patients the L-arginine supplementation increases both the nitrite plasma concentration as well as the HbF synthesis, suggesting the beneficial effects of •NO on the erythroid progenitor cells [49]. Finally, •NO-derived metabolite nitrite is also consumed by the heme-containing myeloperoxidase (MPO). This enzyme, localized within neutrophils and released upon cell activation, catalytically reacts with nitrite in the presence of H2O2, generating powerful radical intermediates, such as nitrogen dioxide (•NO2), which can oxidize and nitrate protein tyrosine residues [50]. MPO also could contribute to the pulmonary hypertension and acute chest syndrome in SCD, since elevated MPO immunoreactivity has been measured in the alveolar epithelium of lung tissues from patients with SCD [51].3. Antioxidant Therapy for SCD
The research of suitable compounds able to (i) limit the hemoglobin-dependent oxidative reactions, (ii) scavenge reactive oxidizing species released and (iii) repair the reactive oxidizing species-mediated tissue oxidative damage is a fundamental step in the clinical management of SCD. There are currently two types of treatment for SCD: primary treatments, which treat the root causes of the disease (gene therapy and HbF inducers antisickling agents) and secondary treatments, which target one of the downstream sequelae of HbS polymerization. In the secondary type of treatment, beside factors working against adhesion, inflammation and thrombophilia, the antioxidant therapy plays an important role for SCD treatment [156][52]. In fact, as already mentioned, oxidative stress can lead to disturbance of cell membranes, exposure of adhesion molecules and damage to the contents of the sickle red blood cells [157][53]. In Table 21 are reported the most promising antioxidant therapeutic strategies, which showed a benefit either in the reducing oxidative stress parameters or in the prevention of pathophysiologic events in SCD patients.3.1. L-Glutamine
L- Glutamine is a precursor of nicotinamide adenine dinucleotide (NAD) required for antioxidant mechanism through the formation of reduced nicotinamide adenine dinucleotide (NADH) [156][52]. Oral administration of L-glutamine in SCD patients has been approved on July 2017 by Food and Drug Administration (FDA). The RBC oxidative damage is most likely consequence of instability of HbS, increasing in free radical generation and impairing antioxidant defenses. This hemoglobin instability leads to denaturation of HbS, through its oxidation to methemoglobin. Methemoglobin reductases slow down this process using NADH. In sickle RBC, there is a decreased NADH/NAD ratio with a consequent decreased NAD redox potential, manifestation of a compensatory mechanism against increased oxidant sensitivity [134][54]. Several trials demonstrated the beneficial effects of oral administration of L-glutamine in SCD in improving cellular redox potential and facilitating protein and glutathione synthesis [157,158,159][53][55][56]. Finally, additional studies suggested that oral L-glutamine supplementation improves the endothelial adhesion of sickle RBC, one of the major factors involved in the pathophysiology of vessel-occlusion. The mechanism underlying this effect is still unclear; however, the improvement of NAD redox potential may protect RBC from oxidant damage and the consequent stimulation of inflammation and expression of adhesion molecules [158][55].| Antioxidants | Mechanisms | Effects in SCD Patients | Comments | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| L- Glutamine | Acts through the formation of reduced NADH. | Improves cellular redox potential and adhesion of sickle RBCs to the endothelium; facilitates protein and glutathione synthesis. | FDA approved; [155,156] | FDA approved; [52][57] | ||||||
| N-Acetylcysteine | Substrate for GSH generation. | Reduces the number of RBCs expressing phosphatidylserine, marker of peroxidative damage to inner membrane of RBCs. | NCT01800526 phase 2 trial; NCT01849016 phase 3 trial | |||||||
| Zinc supplementation | Zinc deficiency is associated with high incidence of infections, vaso-occlusion events and the chronic oxidative stress. | In young SCD patients improves linear growth and weight gain. Has beneficial effects on immunity, inflammatory state and oxidative stress. | NS; [159,160] | NS; [56][58] | ||||||
| Nitric oxide | Reduced | • | NO concentration can be associated to increased levels of free O | 2•− | . | Inhaled | • | NO improves tissue oxygenation and reduces pain in SCD patients with pulmonary hypertension. | NCT00094887 phase 2 trial | |
| L-arginine | Induces GSH synthesis. | Improves | • | NO bioavailability. | NCT02447874 phase 2 trial | |||||
| Alfa-lipoic acid | Induces GSH synthesis. | Increases glutathione level. | NCT01054768 phase 2 trial | |||||||
| L-acetyl-L-carnitine | Improves mitochondrial metabolism, facilitating entry of long-chain fatty acids into mitochondria and decreasing lipid peroxidation in tissue. | Decreases lipid peroxidation. | NCT01054768 phase 2 trial | |||||||
| Gum Arabic | Acts as immuno-modulatory. | Increases total antioxidant capacity and decreased MDA and H | 2 | O | 2 | levels. | NCT04191213 phase 2 trial | |||
| Omega-3 fatty acids | O3FA deficiency correlates with an increase in plasma levels of the inflammatory biomarker | Have beneficial effects on vascular activation, inflammation and antioxidant systems. | [156,157,158,159,160,161,162,163,164, | [55][56][58 | 165, | ] | 166,167, | [59] | 168,169,170] | [52][53][60][61][62][63][64][65][66][67][68] |
| Curcumin | Can modulate the activity of enzymes active in the neutralization of free radicals and it can inhibit ROS-generating enzymes. | Mitigates the effects of iron induced oxidative stress on lipid peroxidation and | • | NO levels. | [169,170] | [67][68] | ||||
| Vitamins A, C and E | Their deficiency increases susceptibility to infection and hemolysis. | Conflicting results about the effectiveness of their supplementation on oxidative stress. | NCT03903133 phase 4 trial | |||||||
| Iron chelators | Avoids excessive iron overload and the consequent ROS generation. | Have a central role in the treatment of transfusion-dependent hemoglobinopathy. | NS; [158] | NS; [55] |
3.2. N-Acetylcysteine (NAC)
NAC is an important antioxidant with pleiotropic effects on inflammation and vasomotor function [156,160][52][58]. It is a substrate for the synthesis of GSH, one of the most important intracellular antioxidants and may play an important role as antioxidant treatment. Indeed, within the cytoplasm, NAC is converted to L-Cysteine, which is a precursor of GSH resulting in an increase of its concentration. GSH has been found to be 32–36% lower in RBCs from SCD patients compared to healthy controls, while some antioxidant enzymes involved in oxidant detoxification, such as SOD and Gpx, have been found significantly higher in patients with SCD [28]. In sickle cell, there is an increased consumption of GSH due to excessive reactive oxidizing species formation, resulting in a decreased ration between GSH and its oxidized form GSSG. In an open label randomized pilot study, Nur and colleagues [135][69] observed that NAC treatment reduced oxidative stress. In particular, they observed (i) a reduced cell membrane phosphatidylserine expression, marker of peroxidative damage to the erythrocyte inner membrane, and (ii) a decrease of AGEs and cell-free hemoglobin. The association between AGEs and the degree of hemolysis and organ complication in sickle cell patients and, on the other hand, an inverse correlation with GSH levels has been recently demonstrated. These results probably suggest that, enhancing GSH levels, NAC treatment could reduce AGEs levels and oxidative tissue damage [135][69].3.3. Zinc Supplementation
Zinc deficiency has been implicated in SCD pathological events. Thus, this element as a therapeutic agent may be very useful in these patients. Zinc deficiency was associated with high incidence of infections connected to weakened cell mediated immunity, with vaso-occlusion events correlated with high level of endothelial cell VCAM-1 molecule and with the chronic oxidative stress. In a study of prepubertal children with SCD-SS, the therapeutic effect of zinc supplementation was evaluated on growth and body composition. Results demonstrated that young SCD patients can benefit from zinc supplementation to improve linear growth and weight gain [161][59]. Furthermore, in a very important double-blind, placebo-controlled study, zinc supplementation (with 25mg elemental zinc as acetate, three times a day for 3 months) ameliorated several pathophysiological parameters chronically existent in these patients: Beneficial effects were observed on immunity, inflammatory state and oxidative stress [162][60].3.4. Nitric Oxide and L-Arginine
In addition to its role in vascular tone, blood flow and adhesion, •NO is known to possess antioxidant properties. Moreover, inhalation of exogenous •NO, can be used to reduce pain in SCD patients with pulmonary hypertension [86][70] and microvascular occlusion in different parts of the body, but controversial results are reported by randomized trials (NCT00094887) [163][61]. In SCD patients, •NO concentration declines, and its reduced bioavailability can be associated to increased levels of free O2•−. Therapy with L-arginine has been demonstrated to improve •NO bioavailability either in transgenic knockout sickle mice or in SCD patients, although in different clinical trials, conflicting results were obtained [164,165][62][63]. The cause of this result has in part been attributed to a deficiency of R-BH4 action. This is an endogenous pterin widely distributed in mammalian tissues that works as a cofactor of aromatic amino acid hydroxylases and nitric oxide synthases. In SCD patients, its deficit is implicated in the mechanism of several diseases such as atherosclerosis, hypertension, diabetic vascular disease and vascular complications [166][64].3.5. α-Lipoic Acid and Acetyl-L-Carnitine
Other compounds with antioxidant properties are α-lipoic acid (LA) and acetyl-L-carnitine (ALCAR). LA increases glutathione level, whereas ALCAR decreases lipid peroxidation [156][52]. One of the mechanisms of antioxidant protection by LA is the induction of GSH synthesis, with a dose-related mechanism, through inducing Nrf2-dependent transcription of γ-glutamyl cysteine ligase (GCL), the rate-controlling enzyme in the synthesis of GSH [167,168][65][66]. On the contrary, the reason of the beneficial effect of ALCAR is not so clear; however, this might occur from improved mitochondrial metabolism, facilitating entry of long-chain fatty acids into mitochondria and decreasing lipid peroxidation in tissue. Studies suggest that this nutrient may be able to maintain the normal shape of RBCs and decrease peroxidative damage [160,168][58][66]. Finally, the evaluation of oxidative stress in human fibroblast exposed to iron excess shows an increased antioxidant effect of combination treatment with LA and ALCAR, suggesting a synergic influence of two compounds [168][66].3.6. Other Antioxidant Agents
Gum Arabic (GA), omega-3 fatty acids and curcumin are reported to diminish oxidative stress in SCD, but their role has not been widely accepted [156][52]. Oral intake of GA has been shown to provide several health benefits, such as probiotic, immuno-modulatory, antioxidant and cytoprotective effects. Available experimental data show its protection against hepatic, renal and cardiac toxicities in rats and, due to its antioxidant properties, this compound may find clinical application sickle cell anemia. In a phase II trial, Kaddam and colleagues treated 47 SCD patients with 30 g/day GA for 12 weeks and demonstrated that GA significantly increased total anti-oxidant capacity and decreased MDA and H2O2-related oxidative markers [169][67]. Limited studies dealing with Ω-3 dietary supplementation are available. Kalish demonstrated the impact of ω-3 fatty acids on vascular activation, inflammation, and antioxidant systems. Authors assessed a modified red cell membrane composition (lower ω -6/ ω -3 ratio), a reduction of neutrophil count and beneficial effects on the cardiovascular system [170][68]. Curcumin could mitigate the effects of iron induced oxidative stress on lipid peroxidation and •NO levels, as showed in rats exposed to iron overloaded toxicity [171,172][71][72]. Despite the observed increased susceptibility to infection and hemolysis in SCD patients with deficiency of vitamins A, C and E, conflicting results are available about the effectiveness of their supplementation on oxidative stress in these patients. Finally, the role of iron chelators (deferiprone, deferoxamine and deferasirox) is central in the treatment of transfusion-dependent hemoglobinopathy, avoiding excessive labile iron overload and the consequent ROS generation [160][58]. Both in in vitro and in animal models, it has been shown that these compounds (i) decrease the RBC membrane-oxidative damage and the production of lipid oxidation product (deferiprone) and (ii) attenuate blood cell adhesion to endothelial cerebral venules (deferoxamine) [160][58]. Finally, in a longitudinal study, SCD patients receiving simultaneously deferasirox and hydroxyurea showed a marked decrease of plasma lipid peroxidation products as well as increased antioxidant capacity levels [173][73]. These promising results should encourage the development of the future research focused on the antioxidant therapy also in combination with the drug treatments. This therapeutic strategy targeted to the reactive oxidizing species-releasing pathway and limiting the intra- and extra-cellular oxidative damage, could reduce the clinical complications of the disease with particular regard to SCD-associated vasculopathy.References
- Chaturvedi, S.; DeBaun, M.R. Evolution of sickle cell disease from a life-threatening disease of children to a chronic disease of adults: The last 40 years. Am. J. Hematol. 2016, 91, 5–14.
- Thein, M.S.; Igbineweka, N.E.; Thein, S.L. Sickle cell disease in the older adult. Pathology 2017, 49, 1–9.
- Aygun, B.; Odame, I. A global perspective on sickle cell disease. Pediatr. Blood Cancer 2012, 59, 386–390.
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat. Rev. Dis. Primers 2018, 4, 18010.
- Piel, F.B.; Steinberg, M.H.; Rees, D.C. Sickle Cell Disease. N. Engl. J. Med. 2017, 377, 305.
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031.
- Ashley-Koch, A.; Yang, Q.; Olney, R.S. Sickle hemoglobin (HbS) allele and sickle cell disease: A HuGE review. Am. J. Epidemiol. 2000, 151, 839–845.
- Stuart, M.J.; Nagel, R.L. Sickle-cell disease. Lancet 2004, 364, 1343–1360.
- Tarasev, M.; Muchnik, M.; Light, L.; Alfano, K.; Chakraborty, S. Individual variability in response to a single sickling event for normal, sickle cell, and sickle trait erythrocytes. Transl. Res. 2017, 181, 96–107.
- Chirico, E.N.; Pialoux, V. Role of oxidative stress in the pathogenesis of sickle cell disease. IUBMB Life 2012, 64, 72–80.
- Dosunmu-Ogunbi, A.M.; Wood, K.C.; Novelli, E.M.; Straub, A.C. Decoding the role of SOD2 in sickle cell disease. Blood Adv. 2019, 3, 2679–2687.
- Steinberg, M.H.; Sebastiani, P. Genetic modifiers of sickle cell disease. Am. J. Hematol. 2012, 87, 795–803.
- Ware, R.E.; de Montalembert, M.; Tshilolo, L.; Abboud, M.R. Sickle cell disease. Lancet 2017, 390, 311–323.
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462.
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183.
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848.
- Ray, D.; Deshmukh, P.; Goswami, K.; Garg, N. Antioxidant vitamin levels in sickle cell disorders. Natl. Med. J. India 2007, 20, 11–13.
- Biswal, S.; Rizwan, H.; Pal, S.; Sabnam, S.; Parida, P.; Pal, A. Oxidative stress, antioxidant capacity, biomolecule damage, and inflammation symptoms of sickle cell disease in children. Hematology 2019, 24, 1–9.
- Al-Naama, L.M.; Hassan, M.K.; Mehdi, J.K. Association of erythrocytes antioxidant enzymes and their cofactors with markers of oxidative stress in patients with sickle cell anemia. Qatar Med. J. 2015, 2015, 14.
- Cardenes, N.; Corey, C.; Geary, L.; Jain, S.; Zharikov, S.; Barge, S.; Novelli, E.M.; Shiva, S. Platelet bioenergetic screen in sickle cell patients reveals mitochondrial complex V inhibition, which contributes to platelet activation. Blood 2014, 123, 2864–2872.
- Wood, K.C.; Hebbel, R.P.; Granger, D.N. Endothelial cell NADPH oxidase mediates the cerebral microvascular dysfunction in sickle cell transgenic mice. FASEB J. 2005, 19, 989–991.
- Wood, K.C.; Hebbel, R.P.; Lefer, D.J.; Granger, D.N. Critical role of endothelial cell-derived nitric oxide synthase in sickle cell disease-induced microvascular dysfunction. Free Radic. Biol. Med. 2006, 40, 1443–1453.
- Wood, K.C.; Granger, D.N. Sickle cell disease: Role of reactive oxygen and nitrogen metabolites. Clin. Exp. Pharmacol. Physiol. 2007, 34, 926–932.
- George, A.; Pushkaran, S.; Konstantinidis, D.G.; Koochaki, S.; Malik, P.; Mohandas, N.; Zheng, Y.; Joiner, C.H.; Kalfa, T.A. Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood 2013, 121, 2099–2107.
- Aslan, M.; Ryan, T.M.; Adler, B.; Townes, T.M.; Parks, D.A.; Thompson, J.A.; Tousson, A.; Gladwin, M.T.; Patel, R.P.; Tarpey, M.M.; et al. Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proc. Natl. Acad. Sci. USA 2001, 98, 15215–15220.
- Schaer, D.J.; Buehler, P.W.; Alayash, A.I.; Belcher, J.D.; Vercellotti, G.M. Hemolysis and free hemoglobin revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 2013, 121, 1276–1284.
- Rifkind, J.M.; Mohanty, J.G.; Nagababu, E. The pathophysiology of extracellular hemoglobin associated with enhanced oxidative reactions. Front. Physiol. 2014, 5, 500.
- Gizi, A.; Papassotiriou, I.; Apostolakou, F.; Lazaropoulou, C.; Papastamataki, M.; Kanavaki, I.; Kalotychou, V.; Goussetis, E.; Kattamis, A.; Rombos, I.; et al. Assessment of oxidative stress in patients with sickle cell disease: The glutathione system and the oxidant-antioxidant status. Blood Cells Mol. Dis. 2011, 46, 220–225.
- Kassa, T.; Jana, S.; Strader, M.B.; Meng, F.; Jia, Y.; Wilson, M.T.; Alayash, A.I. Sickle Cell Hemoglobin in the Ferryl State Promotes betaCys-93 Oxidation and Mitochondrial Dysfunction in Epithelial Lung Cells (E10). J. Biol. Chem. 2015, 290, 27939–27958.
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O., 3rd; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389.
- Berlett, B.S.; Stadtman, E.R. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 1997, 272, 20313–20316.
- Mano, J. Reactive carbonyl species: Their production from lipid peroxides, action in environmental stress, and the detoxification mechanism. Plant. Physiol. Biochem. 2012, 59, 90–97.
- Azarov, I.; He, X.; Jeffers, A.; Basu, S.; Ucer, B.; Hantgan, R.R.; Levy, A.; Kim-Shapiro, D.B. Rate of nitric oxide scavenging by hemoglobin bound to haptoglobin. Nitric Oxide 2008, 18, 296–302.
- Nielsen, M.J.; Moller, H.J.; Moestrup, S.K. Hemoglobin and heme scavenger receptors. Antioxid. Redox Signal. 2010, 12, 261–273.
- Koskenkorva-Frank, T.S.; Weiss, G.; Koppenol, W.H.; Burckhardt, S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: Insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic. Biol. Med. 2013, 65, 1174–1194.
- Balla, J.; Jacob, H.S.; Balla, G.; Nath, K.; Eaton, J.W.; Vercellotti, G.M. Endothelial-cell heme uptake from heme proteins: Induction of sensitization and desensitization to oxidant damage. Proc. Natl. Acad. Sci. USA 1993, 90, 9285–9289.
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188.
- Belcher, J.D.; Beckman, J.D.; Balla, G.; Balla, J.; Vercellotti, G. Heme degradation and vascular injury. Antioxid. Redox Signal. 2010, 12, 233–248.
- Aslan, M.; Freeman, B.A. Oxidant-mediated impairment of nitric oxide signaling in sickle cell disease—Mechanisms and consequences. Cell Mol. Biol. 2004, 50, 95–105.
- Jeffers, A.; Gladwin, M.T.; Kim-Shapiro, D.B. Computation of plasma hemoglobin nitric oxide scavenging in hemolytic anemias. Free Radic. Biol. Med. 2006, 41, 1557–1565.
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Investig. 2017, 127, 750–760.
- Shiva, S.; Wang, X.; Ringwood, L.A.; Xu, X.; Yuditskaya, S.; Annavajjhala, V.; Miyajima, H.; Hogg, N.; Harris, Z.L.; Gladwin, M.T. Ceruloplasmin is a NO oxidase and nitrite synthase that determines endocrine NO homeostasis. Nat. Chem. Biol. 2006, 2, 486–493.
- Grubina, R.; Basu, S.; Tiso, M.; Kim-Shapiro, D.B.; Gladwin, M.T. Nitrite reductase activity of hemoglobin S (sickle) provides insight into contributions of heme redox potential versus ligand affinity. J. Biol. Chem. 2008, 283, 3628–3638.
- Rees, D.C.; Gibson, J.S. Biomarkers in sickle cell disease. Br. J. Haematol. 2012, 156, 433–445.
- Antwi-Boasiako, C.; Dankwah, G.B.; Aryee, R.; Hayfron-Benjamin, C.; Aboagye, G.; Campbell, A.D. Correlation of lipid peroxidation and nitric oxide metabolites, trace elements, and antioxidant enzymes in patients with sickle cell disease. J. Clin. Lab. Anal. 2020, 34, e23294.
- Reiter, C.D.; Gladwin, M.T. An emerging role for nitric oxide in sickle cell disease vascular homeostasis and therapy. Curr. Opin. Hematol. 2003, 10, 99–107.
- Azizi, E.; Dror, Y.; Wallis, K. Arginase activity in erythrocytes of healthy and ill children. Clin. Chim Acta 1970, 28, 391–396.
- Morris, C.R.; Kato, G.J.; Poljakovic, M.; Wang, X.; Blackwelder, W.C.; Sachdev, V.; Hazen, S.L.; Vichinsky, E.P.; Morris, S.M., Jr.; Gladwin, M.T. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA 2005, 294, 81–90.
- Elias, D.B.; Barbosa, M.C.; Rocha, L.B.; Dutra, L.L.; Silva, H.F.; Martins, A.M.; Goncalves, R.P. L-arginine as an adjuvant drug in the treatment of sickle cell anaemia. Br. J. Haematol. 2013, 160, 410–412.
- Davies, M.J.; Hawkins, C.L. The Role of Myeloperoxidase in Biomolecule Modification, Chronic Inflammation, and Disease. Antioxid. Redox Signal. 2020, 32, 957–981.
- Baldus, S.; Eiserich, J.P.; Brennan, M.L.; Jackson, R.M.; Alexander, C.B.; Freeman, B.A. Spatial mapping of pulmonary and vascular nitrotyrosine reveals the pivotal role of myeloperoxidase as a catalyst for tyrosine nitration in inflammatory diseases. Free Radic. Biol. Med. 2002, 33, 1010.
- Kapoor, S.; Little, J.A.; Pecker, L.H. Advances in the Treatment of Sickle Cell Disease. Mayo Clin. Proc. 2018, 93, 1810–1824.
- Niihara, Y.; Macan, H.; Eckman, J.R.; Koh, H.; Cooper, M.L.; Ziegler, T.R.; Razon, R.; Tanaka, K.R.; Stark, C.W.; Johnson, C.S. L-Glutamine Therapy Reduces Hospitalization for Sickle Cell Anemia and Sickle β°-Thalassemia Patients at Six Months—A Phase II Randomized Trial. Clin. Pharmacol. Biopharm. 2014, 3, 116.
- Zerez, C.R.; Lachant, N.A.; Lee, S.J.; Tanaka, K.R. Decreased erythrocyte nicotinamide adenine dinucleotide redox potential and abnormal pyridine nucleotide content in sickle cell disease. Blood 1988, 71, 512–515.
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N. Engl. J. Med. 2018, 379, 226–235.
- Ortiz de Montellano, P.R. A New Step in the Treatment of Sickle Cell DiseasePublished as part of the Biochemistry series “Biochemistry to Bedside”. Biochemistry 2018, 57, 470–471.
- Vendrame, F.; Olops, L.; Saad, S.T.O.; Costa, F.F.; Fertrin, K.Y. Differences in heme and hemopexin content in lipoproteins from patients with sickle cell disease. J. Clin. Lipidol. 2018, 12, 1532–1538.
- Silva, D.G.H.; Belini Junior, E.; de Almeida, E.A.; Bonini-Domingos, C.R. Oxidative stress in sickle cell disease: An overview of erythrocyte redox metabolism and current antioxidant therapeutic strategies. Free Radic. Biol. Med. 2013, 65, 1101–1109.
- Zemel, B.S.; Kawchak, D.A.; Fung, E.B.; Ohene-Frempong, K.; Stallings, V.A. Effect of zinc supplementation on growth and body composition in children with sickle cell disease. Am. J. Clin. Nutr. 2002, 75, 300–307.
- Bao, B.; Prasad, A.S.; Beck, F.W.; Snell, D.; Suneja, A.; Sarkar, F.H.; Doshi, N.; Fitzgerald, J.T.; Swerdlow, P. Zinc supplementation decreases oxidative stress, incidence of infection, and generation of inflammatory cytokines in sickle cell disease patients. Transl. Res. 2008, 152, 67–80.
- Aboursheid, T.; Albaroudi, O.; Alahdab, F. Inhaled nitric oxide for treating pain crises in people with sickle cell disease. Cochrane Database Syst. Rev. 2019, 10, CD011808.
- Dasgupta, T.; Hebbel, R.P.; Kaul, D.K. Protective effect of arginine on oxidative stress in transgenic sickle mouse models. Free Radic. Biol. Med. 2006, 41, 1771–1780.
- Kaul, D.K.; Zhang, X.; Dasgupta, T.; Fabry, M.E. Arginine therapy of transgenic-knockout sickle mice improves microvascular function by reducing non-nitric oxide vasodilators, hemolysis, and oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H39–H47.
- Vasquez-Vivar, J. Tetrahydrobiopterin, superoxide, and vascular dysfunction. Free Radic. Biol. Med. 2009, 47, 1108–1119.
- Lal, A.; Suh, J.H.; Atamna, W.; Canty, B.; Hagar, W.; Vichinsky, E.F.; Ames, B. Anti-oxidant treatment with alipoic acid and acetyl L-carnitine in hemoglobinopathies. Blood 2007, 11, 3799.
- Lal, A.; Atamna, W.; Killilea, D.W.; Suh, J.H.; Ames, B.N. Lipoic acid and acetyl-carnitine reverse iron-induced oxidative stress in human fibroblasts. Redox Rep. 2008, 13, 2–10.
- Kaddam, L.; Fadl-Elmula, I.; Eisawi, O.A.; Abdelrazig, H.A.; Salih, M.A.; Lang, F.; Saeed, A.M. Gum Arabic as novel anti-oxidant agent in sickle cell anemia, phase II trial. BMC Hematol. 2017, 17, 4.
- Kalish, B.T.; Matte, A.; Andolfo, I.; Iolascon, A.; Weinberg, O.; Ghigo, A.; Cimino, J.; Siciliano, A.; Hirsch, E.; Federti, E.; et al. Dietary omega-3 fatty acids protect against vasculopathy in a transgenic mouse model of sickle cell disease. Haematologica 2015, 100, 870–880.
- Nur, E.; Brandjes, D.P.; Teerlink, T.; Otten, H.M.; Oude Elferink, R.P.; Muskiet, F.; Evers, L.M.; Ten Cate, H.; Biemond, B.J.; Duits, A.J.; et al. N-acetylcysteine reduces oxidative stress in sickle cell patients. Ann. Hematol. 2012, 91, 1097–1105.
- Potoka, K.P.; Gladwin, M.T. Vasculopathy and pulmonary hypertension in sickle cell disease. Am. J. Physiol. Lung Cell. Mol. Physiol 2015, 308, L314–L324.
- Badria, F.A.; Ibrahim, A.S.; Badria, A.F.; Elmarakby, A.A. Curcumin Attenuates Iron Accumulation and Oxidative Stress in the Liver and Spleen of Chronic Iron-Overloaded Rats. PLoS ONE 2015, 10, e0134156.
- Badria, F.A.; Ibrahim, A.S.; Badria, A.F.; Elmarakby, A.A. Correction: Curcumin attenuates iron accumulation and oxidative stress in the liver and spleen of chronic iron-overloaded rats. PLoS ONE 2020, 15, e0243398.
- Belini Junior, E.; da Silva, D.G.; Torres Lde, S.; de Almeida, E.A.; Cancado, R.D.; Chiattone, C.; Bonini-Domingos, C.R. Oxidative stress and antioxidant capacity in sickle cell anaemia patients receiving different treatments and medications for different periods of time. Ann. Hematol. 2012, 91, 479–489.