 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Efrosyni Paraskeva | -- | 2340 | 2022-05-20 12:07:34 | | | |
| 2 | Jessie Wu | Meta information modification | 2340 | 2022-05-23 06:37:03 | | | | |
| 3 | Jessie Wu | Meta information modification | 2340 | 2022-05-23 08:46:57 | | |
Video Upload Options
Oxygen deprivation or hypoxia characterizes a number of serious pathological conditions and elicits a number of adaptive changes that are mainly mediated at the transcriptional level by the family of hypoxia-inducible factors (HIFs). The HIF target gene repertoire includes genes responsible for the regulation of metabolism, oxygen delivery and cell survival. Although the involvement of HIFs in the regulation of carbohydrate metabolism and the switch to anaerobic glycolysis under hypoxia is well established, their role in the control of lipid anabolism and catabolism remains still relatively obscure. Recent evidence indicates that many aspects of lipid metabolism are modified during hypoxia or in tumor cells in a HIF-dependent manner, contributing significantly to the pathogenesis and/or progression of cancer and metabolic disorders.
1. The Involvement of Hypoxia-Inducible Factors in the Regulation of Lipid Metabolism
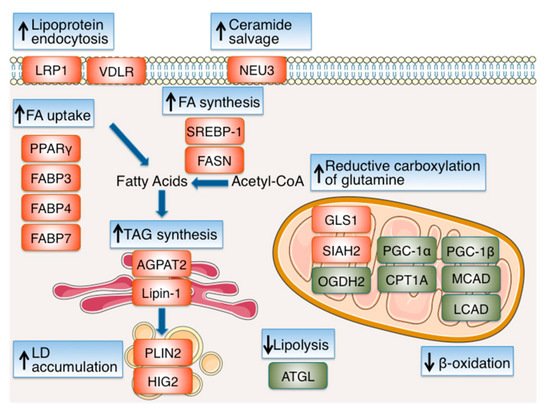
| Functional Category /Protein Name |
HIF Isoform & Effect | Outcome & Experimental Evidence | Ref. |
|---|---|---|---|
| FA & Lipoprotein Uptake | |||
| PPARγ | HIF-1 Positive | Increased expression HIF-1 binds to the promoter of PPARγ and activates its transcription |
[6] |
| FABP3 | HIF-1 Positive | Increased expression HIF-1α depletion inhibits the induction of FABP3 under hypoxia |
[7] |
| FABP4 | HIF-1 Positive | Increased expression HIF-1 binds to the promoter of FABP4 and activates its transcription |
[8] |
| FABP7 | HIF-1 Positive | Increased expression HIF-1α depletion inhibits the induction of FABP7 under hypoxia |
[7] |
| LRP1 | HIF-1 Positive | Increased expression HIF-1α binds to the LRP1 promoter and activates its transcription |
[9] |
| VDLR | HIF-1 Positive | Increased expression HIF-1α depletion inhibits activation of VDLR promoter under hypoxia |
[10] |
| Reductive Carboxylation of Glutamine | |||
| GLS1 | HIF-1 Positive | Increased expression HIF-1α depletion inhibits the induction of GLS1 under hypoxia |
[16] |
| OGDH2 | HIF-1 Negative | Increased proteolysis SIAH2 (a HIF-1 target) mediates proteolysis of OGDH2 |
[15] |
| Ceramide Salvage | |||
| NEU3 | HIF-2 Positive | Increased expression HIF-2α binds to the NEU3 promoter and activates its transcription |
[30] |
| FA Synthesis | |||
| SREBP-1 | HIF-1 Positive | Up-regulation Inhibition of HIF-1 impairs phospho-SREBP-1 increase under hypoxia |
[17][27] |
| FASN | HIF-1 Positive | Increased expression Inhibition of HIF-1 impairs the induction of FASN under hypoxia Increased binding of SREBP-1 to the FASN promoter under hypoxia |
[17] |
| TG Synthesis | |||
| AGPAT2 | HIF-1 Positive | Increased expression HIF-1 binds to the promoter of AGPAT2 and activates its transcription |
[20] |
| Lipin-1 | HIF-1 Positive | Increased expression HIF-1 binds to the promoter of LPIN1 and activates its transcription |
[21] |
| LD Accumulation | |||
| PLIN2 | HIF-2 Positive | Increased expression HIF-2α depletion inhibits the induction of PLIN2 under hypoxia |
[23] |
| HIG2 | HIF-1 Positive | Increased expression HIF-1 binds to the promoter of HIG2 and activates its transcription |
[24] |
| β-Oxidation | |||
| PGC-1α | HIF-1 & HIF-2 Negative | Reduced expression HIF-1α or HIF-2α depletion inhibits reduction of PGC-1α expression under hypoxia |
[27] |
| CPT1A | HIF-1 & HIF-2 Negative | Reduced expression HIF-1α or HIF-2α depletion inhibit reduction of CPT1A expression under hypoxia |
[27][28] |
| MCAD | HIF-1 Negative | Reduced expression HIF-1α depletion inhibits reduction of MCAD expression under hypoxia |
[29] |
| LCAD | HIF-1 Negative | Reduced expression HIF-1α depletion inhibits reduction of LCAD expression under hypoxia |
[29] |
| PGC-1β | HIF-1 Negative | Reduced expression HIF-1α depletion inhibits reduction of PGC-1β expression under hypoxia |
[29] |
2. Hypoxia-Inducible Factors-Dependent Regulation of Lipid Metabolism in Cardiovascular Disease
References
- Samanta, D.; Semenza, G.L. Metabolic adaptation of cancer and immune cells mediated by hypoxia-inducible factors. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 15–22.
- Xie, H.; Simon, M.C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 2017, 292, 16825–16832.
- Kim, W.Y.; Safran, M.; Buckley, M.R.; Ebert, B.L.; Glickman, J.; Bosenberg, M.; Regan, M.; Kaelin, W.G., Jr. Failure to prolyl hydroxylate hypoxia-inducible factor alpha phenocopies VHL inactivation in vivo. EMBO J. 2006, 25, 4650–4662.
- Zhang, L.; Li, L.; Liu, H.; Prabhakaran, K.; Zhang, X.; Borowitz, J.L.; Isom, G.E. HIF-1alpha activation by a redox-sensitive pathway mediates cyanide-induced BNIP3 upregulation and mitochondrial-dependent cell death. Free Radic. Biol. Med. 2007, 43, 117–127.
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197.
- Krishnan, J.; Suter, M.; Windak, R.; Krebs, T.; Felley, A.; Montessuit, C.; Tokarska-Schlattner, M.; Aasum, E.; Bogdanova, A.; Perriard, E.; et al. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009, 9, 512–524.
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365.
- Hu, B.; Guo, Y.; Garbacz, W.G.; Jiang, M.; Xu, M.; Huang, H.; Tsung, A.; Billiar, T.R.; Ramakrishnan, S.K.; Shah, Y.M.; et al. Fatty acid binding protein-4 (FABP4) is a hypoxia inducible gene that sensitizes mice to liver ischemia/reperfusion injury. J. Hepatol. 2015, 63, 855–862.
- Castellano, J.; Aledo, R.; Sendra, J.; Costales, P.; Juan-Babot, O.; Badimon, L.; Llorente-Cortes, V. Hypoxia stimulates low-density lipoprotein receptor-related protein-1 expression through hypoxia-inducible factor-1alpha in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1411–1420.
- Perman, J.C.; Bostrom, P.; Lindbom, M.; Lidberg, U.; StAhlman, M.; Hagg, D.; Lindskog, H.; Scharin Tang, M.; Omerovic, E.; Mattsson Hulten, L.; et al. The VLDL receptor promotes lipotoxicity and increases mortality in mice following an acute myocardial infarction. J. Clin. Investig. 2011, 121, 2625–2640.
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388.
- Gameiro, P.A.; Yang, J.; Metelo, A.M.; Perez-Carro, R.; Baker, R.; Wang, Z.; Arreola, A.; Rathmell, W.K.; Olumi, A.; Lopez-Larrubia, P.; et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013, 17, 372–385.
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616.
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384.
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292.
- Xiang, L.; Mou, J.; Shao, B.; Wei, Y.; Liang, H.; Takano, N.; Semenza, G.L.; Xie, G. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. 2019, 10, 40.
- Furuta, E.; Pai, S.K.; Zhan, R.; Bandyopadhyay, S.; Watabe, M.; Mo, Y.Y.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008, 68, 1003–1011.
- Wang, H.; Airola, M.V.; Reue, K. How lipid droplets “TAG” along: Glycerolipid synthetic enzymes and lipid storage. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1131–1145.
- Siniossoglou, S. Phospholipid metabolism and nuclear function: Roles of the lipin family of phosphatidic acid phosphatases. Biochim. Biophys. Acta 2013, 1831, 575–581.
- Triantafyllou, E.A.; Georgatsou, E.; Mylonis, I.; Simos, G.; Paraskeva, E. Expression of AGPAT2, an enzyme involved in the glycerophospholipid/triacylglycerol biosynthesis pathway, is directly regulated by HIF-1 and promotes survival and etoposide resistance of cancer cells under hypoxia. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1142–1152.
- Mylonis, I.; Sembongi, H.; Befani, C.; Liakos, P.; Siniossoglou, S.; Simos, G. Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression. J. Cell Sci. 2012, 125, 3485–3493.
- Kourti, M.; Ikonomou, G.; Giakoumakis, N.N.; Rapsomaniki, M.A.; Landegren, U.; Siniossoglou, S.; Lygerou, Z.; Simos, G.; Mylonis, I. CK1delta restrains lipin-1 induction, lipid droplet formation and cell proliferation under hypoxia by reducing HIF-1alpha/ARNT complex formation. Cell. Signal. 2015.
- Qiu, B.; Ackerman, D.; Sanchez, D.J.; Li, B.; Ochocki, J.D.; Grazioli, A.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; Simon, M.C. HIF2alpha-Dependent Lipid Storage Promotes Endoplasmic Reticulum Homeostasis in Clear-Cell Renal Cell Carcinoma. Cancer Discov. 2015, 5, 652–667.
- Gimm, T.; Wiese, M.; Teschemacher, B.; Deggerich, A.; Schodel, J.; Knaup, K.X.; Hackenbeck, T.; Hellerbrand, C.; Amann, K.; Wiesener, M.S.; et al. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J. 2010, 24, 4443–4458.
- Maier, A.; Wu, H.; Cordasic, N.; Oefner, P.; Dietel, B.; Thiele, C.; Weidemann, A.; Eckardt, K.U.; Warnecke, C. Hypoxia-inducible protein 2 Hig2/Hilpda mediates neutral lipid accumulation in macrophages and contributes to atherosclerosis in apolipoprotein E-deficient mice. FASEB J. 2017, 31, 4971–4984.
- Zhang, X.; Saarinen, A.M.; Hitosugi, T.; Wang, Z.; Wang, L.; Ho, T.H.; Liu, J. Inhibition of intracellular lipolysis promotes human cancer cell adaptation to hypoxia. eLife 2017, 6.
- Liu, Y.; Ma, Z.; Zhao, C.; Wang, Y.; Wu, G.; Xiao, J.; McClain, C.J.; Li, X.; Feng, W. HIF-1α and HIF-2α are critically involved in hypoxia-induced lipid accumulation in hepatocytes through reducing PGC-1α-mediated fatty acid β-oxidation. Toxicol. Lett. 2014, 226, 117–123.
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I.; et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017, 8, 1769.
- Huang, D.; Li, T.; Li, X.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L.; et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014, 8, 1930–1942.
- Xie, C.; Yagai, T.; Luo, Y.; Liang, X.; Chen, T.; Wang, Q.; Sun, D.; Zhao, J.; Ramakrishnan, S.K.; Sun, L.; et al. Activation of intestinal hypoxia-inducible factor 2alpha during obesity contributes to hepatic steatosis. Nat. Med. 2017, 23, 1298–1308.
- Bostrom, P.; Magnusson, B.; Svensson, P.A.; Wiklund, O.; Boren, J.; Carlsson, L.M.; Stahlman, M.; Olofsson, S.O.; Hulten, L.M. Hypoxia converts human macrophages into triglyceride-loaded foam cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1871–1876.
- Lei, L.; Mason, S.; Liu, D.; Huang, Y.; Marks, C.; Hickey, R.; Jovin, I.S.; Pypaert, M.; Johnson, R.S.; Giordano, F.J. Hypoxia-inducible factor-dependent degeneration, failure, and malignant transformation of the heart in the absence of the von Hippel-Lindau protein. Mol. Cell Biol. 2008, 28, 3790–3803.
- Lin, Q.; Huang, Y.; Booth, C.J.; Haase, V.H.; Johnson, R.S.; Celeste Simon, M.; Giordano, F.J.; Yun, Z. Activation of hypoxia-inducible factor-2 in adipocytes results in pathological cardiac hypertrophy. J. Am. Heart Assoc. 2013, 2, e000548.
- Marsch, E.; Demandt, J.A.; Theelen, T.L.; Tullemans, B.M.; Wouters, K.; Boon, M.R.; van Dijk, T.H.; Gijbels, M.J.; Dubois, L.J.; Meex, S.J.; et al. Deficiency of the oxygen sensor prolyl hydroxylase 1 attenuates hypercholesterolaemia, atherosclerosis, and hyperglycaemia. Eur. Heart J. 2016, 37, 2993–2997.
- Rahtu-Korpela, L.; Maatta, J.; Dimova, E.Y.; Horkko, S.; Gylling, H.; Walkinshaw, G.; Hakkola, J.; Kivirikko, K.I.; Myllyharju, J.; Serpi, R.; et al. Hypoxia-Inducible Factor Prolyl 4-Hydroxylase-2 Inhibition Protects Against Development of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 608–617.
- Dai, Z.; Li, M.; Wharton, J.; Zhu, M.M.; Zhao, Y.Y. Prolyl-4 Hydroxylase 2 (PHD2) Deficiency in Endothelial Cells and Hematopoietic Cells Induces Obliterative Vascular Remodeling and Severe Pulmonary Arterial Hypertension in Mice and Humans Through Hypoxia-Inducible Factor-2alpha. Circulation 2016, 133, 2447–2458.
- Ivan, M.; Kaelin, W.G., Jr. The EGLN-HIF O2-Sensing System: Multiple Inputs and Feedbacks. Mol. Cell 2017, 66, 772–779.
- Shimoda, L.A.; Semenza, G.L. HIF and the lung: Role of hypoxia-inducible factors in pulmonary development and disease. Am. J. Respir. Crit. Care Med. 2011, 183, 152–156.
- Shimoda, L.A.; Laurie, S.S. HIF and pulmonary vascular responses to hypoxia. J. Appl. Physiol. 2014, 116, 867–874.
- D’Alessandro, A.; El Kasmi, K.C.; Plecita-Hlavata, L.; Jezek, P.; Li, M.; Zhang, H.; Gupte, S.A.; Stenmark, K.R. Hallmarks of Pulmonary Hypertension: Mesenchymal and Inflammatory Cell Metabolic Reprogramming. Antioxid. Redox Signal. 2018, 28, 230–250.
- Sutendra, G.; Michelakis, E.D. The metabolic basis of pulmonary arterial hypertension. Cell Metab. 2014, 19, 558–573.
- Sutendra, G.; Bonnet, S.; Rochefort, G.; Haromy, A.; Folmes, K.D.; Lopaschuk, G.D.; Dyck, J.R.; Michelakis, E.D. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci. Transl. Med. 2010, 2, 44ra58.
- Rubin, L.J. Metabolic dysfunction in the pathogenesis of pulmonary hypertension. Cell Metab. 2010, 12, 313–314.
- Izquierdo-Garcia, J.L.; Arias, T.; Rojas, Y.; Garcia-Ruiz, V.; Santos, A.; Martin-Puig, S.; Ruiz-Cabello, J. Metabolic Reprogramming in the Heart and Lung in a Murine Model of Pulmonary Arterial Hypertension. Front. Cardiovasc. Med. 2018, 5, 110.

