Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Matthias Laesche | -- | 5087 | 2022-05-13 08:12:17 | | | |
| 2 | Amina Yu | -35 word(s) | 5052 | 2022-05-16 03:06:51 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Laesche, M.; Gründker, C.; Gallwas, J. Hormonal Stimuli, Leading HPV Infection to Cervical Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/22902 (accessed on 08 August 2026).
Laesche M, Gründker C, Gallwas J. Hormonal Stimuli, Leading HPV Infection to Cervical Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/22902. Accessed August 08, 2026.
Laesche, Matthias, Carsten Gründker, Julia Gallwas. "Hormonal Stimuli, Leading HPV Infection to Cervical Cancer" Encyclopedia, https://encyclopedia.pub/entry/22902 (accessed August 08, 2026).
Laesche, M., Gründker, C., & Gallwas, J. (2022, May 13). Hormonal Stimuli, Leading HPV Infection to Cervical Cancer. In Encyclopedia. https://encyclopedia.pub/entry/22902
Laesche, Matthias, et al. "Hormonal Stimuli, Leading HPV Infection to Cervical Cancer." Encyclopedia. Web. 13 May, 2022.
Copy Citation
The special thing about an HPV infection is that it is not only able to trick the immune system in a sophisticated way, but also, through genetic integration into the host genome, to use all the resources available to the host cells to complete the replication cycle of the virus without activating the alarm mechanisms of immune recognition and elimination. The mechanisms utilized by the virus are the metabolic, immune, and hormonal signaling pathways that it manipulates. Since the virus is dependent on replication enzymes of the host cells, it also intervenes in the cell cycle of the differentiating keratinocytes and shifts their terminal differentiation to the uppermost layers of the squamocolumnar transformation zone (TZ) of the cervix.
cervical cancer
human papillomavirus (HPV)
metabolism
estrogens
1. Introduction
The female reproductive tract is divided into an upper and a lower part. The upper part forms the uterus, the fallopian tubes and the ovaries. The lower part forms the vagina and the cervix. The cervix is divided into the endocervix (cervical canal) and ectocervix, which both can be examined by colposcopy. While the ectocervix is a stratified squamous epithelium, the endocervix consists of columnar epithelium. The squamocolumnar transformation zone (TZ) lies between the endocervix and the ectocervix. The cervix, as a mucosal epithelium, is susceptible to injury from sexual activity, which makes it sensitive to sexually transmitted infections (STIs).
A disease relevant to the cervix is infection with the human papillomavirus (HPV). Due to its reproductive cycle, the virus is dependent on the stratified epithelium of the cervix (Figure 1). High-risk HPV infections are responsible for 99.7% of cervical cancers, more than 90% of anal cancers, 50% of head and neck cancers, 40% of vulva cancers, as well as a minority of cancers of the vagina and penis [1][2][3][4]. HPV represents the most common sexually transmitted infection. The virus is responsible for approximately 5% of all cancers worldwide [5][6][7][8][9][10][11][12].
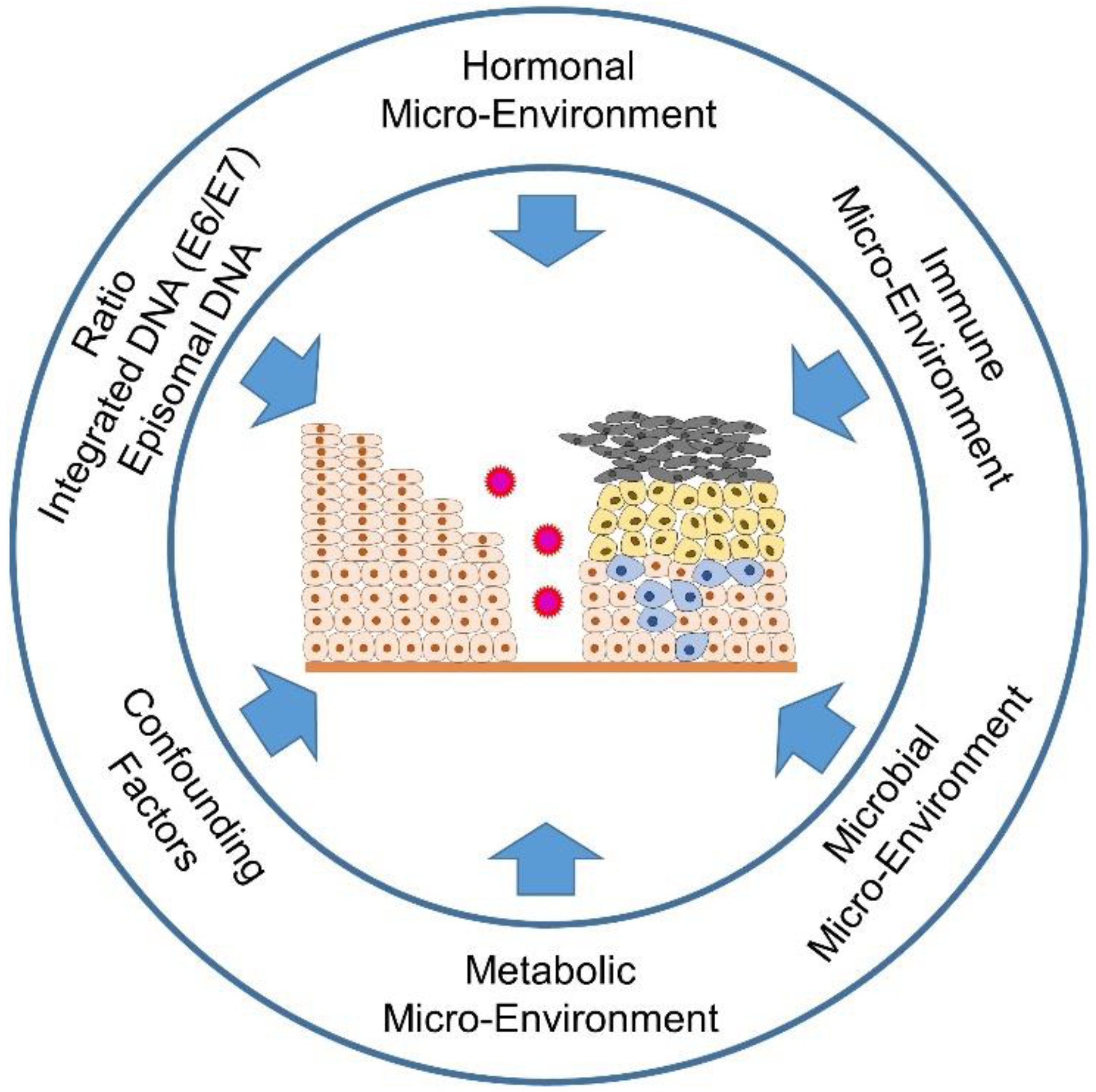
Figure 1. Important influences on cervical carcinogenesis.
It is not only viruses that determine the fate of the affected cells through their development cycle, but also the microenvironment of the affected cells, for example, the surrounding tissue, the immune cells infiltrating the tissue, but also sex steroid hormones such as estrogen and progesterone support the metamorphosis of the developing malignant cells [13][14][15]. Further influences on the fate of the developing malignancy also depend on the epidemiological and socioeconomic background of the patients since not all women have equal access to vaccination and screening programs [16][17]. Other confounding factors include age differences (pre- and postmenopausal), the time of sampling, depending on the menstrual cycle, tobacco smoking and the use of various contraceptives or other concomitant infections, such as Chlamydia trachomatis or human immunodeficiency virus (HIV) infections [18][19][20][21][22]. The crosstalk between the vaginal microbiome, including HPV and other STIs, the estrobolome (capable of metabolizing estrogens via β-glucuronidases and sulfatases) and the non-estrogen-metabolizing tryptophan gut microbiota (melatonin-producing) [23][24][25] will be focuesed. Further, it will be examined the influence of selective estrogen receptor modulators (SERMs) and selective estrogen enzyme modulators (SEEMs) or the effect of aromatase inhibitors (AIs) on estrogen signaling [15][26]. Moreover, it will take a look not only at the supportive component of sex steroid hormones in cervical carcinogenesis, but also at their (perhaps unexpected) option as a therapeutic component in the treatment of cervical lesions [13].
Besides the influences of HPV infection and the accompanying microbiota, immune [15] and metabolic [27] signaling are important determinants of patients’ health fate (pro- and anti-tumorigenic forces of the immune cells infiltrating the lesions). Higher estrogen levels generally have a positive effect on the production of antibodies and may also be associated with the differences in innate pathogen sensing and immune cell populations, thus also having a positive effect on the immune response to an infection [28][29]. On the other hand, HPV is able to suppress the arm of innate immunity via its (onco)proteins (E2, E5, E6 and E7) [30][31][32] The metabolic signaling is also influenced by the chronically altered infection and the subsequent genetic incorporation of the HPV genome coding for the oncoproteins E6 and E7 into the host DNA. Here, aerobic glycolysis (Warburg effect), one of the hallmarks of cancer, is increased in order to create the metabolic basis for increased proliferation, migration and metastasis. The two HPV oncoproteins E6 and E7 work cooperatively in order not only to suppress apoptosis by repressing the tumor-suppressor proteins pRb and p53, but also to increase glycolysis and inhibit oxidative phosphorylation (Figure 2) [27]. It will be addressed that the mechanisms required for this in detail.
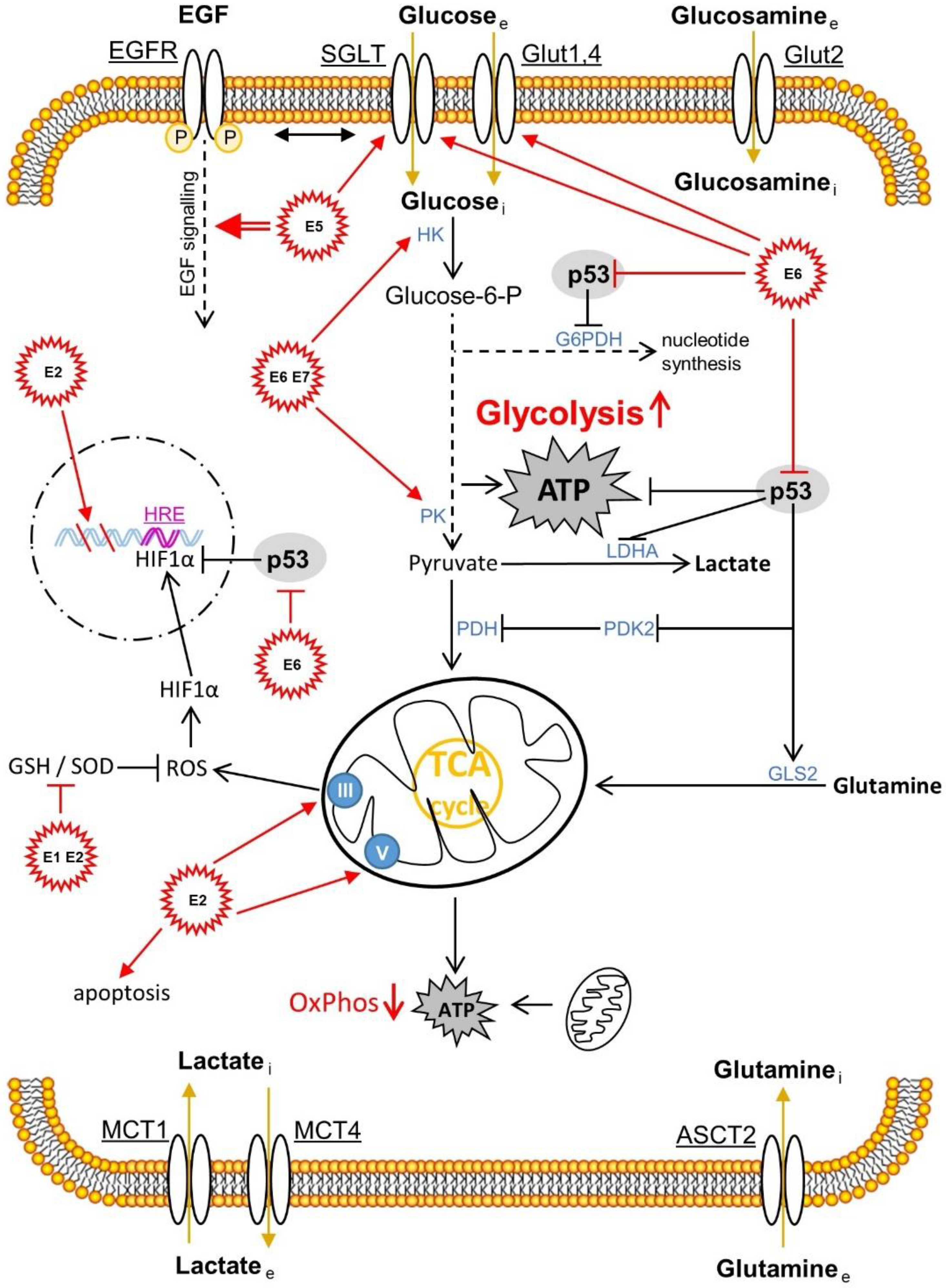
Figure 2. The HPV proteins E1, E2, E5, E6 and E7 and their effects on the metabolism of the transformed cervical tissue. The correlations shown are mainly presented in Section 3.1. ASCT2, amino acid transporter 2; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; G6PDH, glucose-6-phosphat-dehydrogenase; GLS2, glutaminase 2; Glut, glucose transporter; GSH, glutathione; HRE, hypoxia response element; HIF1α, hypoxia-inducible factor 1α; HK, hexokinase; LDHA, lactate dehydrogenase A; MCT, monocarboxylate transporter; OxPhos, oxidative phosphorylation; PDH, pyruvate dehydrogenase; PK, pyruvate kinase; PDK2, pyruvate dehydrogenase kinase isoform 2; ROS, reactive oxygen species; SGLT, sodium glucose transporter; SOD, superoxide dismutase. Reproduced from [30].
2. The Effects of the Hormone Microenvironment on Cervical Carcinogenesis
2.1. General Information about Sex Steroid Hormones, Their Biosynthesis and Their Localization in the Female Body
Of the estrogens circulating in the female body, 17β-estradiol (also referred to as estradiol or E2) is the predominant form of the sex steroid hormone. It plays an important role in both sexes. In addition to the discussed functions in the menstrual cycle, the predominant estrogen is present at high levels during the reproductive phase in women [33]. At the beginning of menopause, the production of estrogen drops significantly and thus leads to well-known symptoms in women in this phase of life [33]. Bone health, heart health, female fertility, various regulatory effects on glucose metabolism, the maintenance of immune function, nerve function and the role of estrogens in the treatment of many pathologies are also modulated by estrogen signaling [33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52]. The estrogen circulating in the woman’s body acts in an endocrine, paracrine or intracrine manner and binds to the three most important of its receptors, the estrogen receptor α (ERα), the estrogen receptor β (ERβ) or the G protein-coupled estrogen receptor 1 (GPER1) [53][54].
Most estrogen is formed in the ovaries of women, but also in adipose tissue and the adrenal glands in women and men [54][55][56][57]. Androstenedione (ASD), which is also well-known in the bodybuilder scene, can be formed in adipose tissue from low-density lipoprotein (LDL) cholesterol via various enzymatic steps. ASD can then be metabolized to any of the steroid hormones in addition to estrogen [54][58][59]. In addition to the gonads (ovaries), estradiol is also synthesized via Peyer’s patches in the intestine [58]. In 1645, Italian anatomist and surgeon Marco Aurelio Severino referred to these structures in the gut as “lymphoid follicles.” Just 32 years later, they were named Peyer’s patches after the Swiss pathologist Johann Conrad Peyer, who described these intestinal structures in detail. The intestines, but also all other human mucous membranes, are colonized by a large number of microorganisms. These symbiotic but also pathogenic microorganisms form, among other things, the intestinal flora (intestinal microbiota), a community of bacteria, primordial bacteria, fungi, protozoa and even viruses [60]. The habitat of the microorganisms, their genome, together with the neighboring microenvironment, are called the microbiome [61]. The number of bacteria colonizing human mucous membranes and other epithelia is estimated to be as large as there are cells in the human body [62]. However, compared to humans, the bacterial community has a metagenome that is 100 times larger [63]. What is special about the intestinal microbiome is that, as a so-called estrobolome [15][64], in contrast to the non-metabolizing tryptophan microbiome, it is able to regulate the free estradiol level in the intestine and in the mucosal epithelia outside the intestine. In addition to estradiol (E2), estrone (E1) is also formed in the human body when androstenedione (ASD) is aromatized. This mainly happens in postmenopausal women. If the woman is pregnant, her body mainly produces the third form of estrogen, estriol (E3) [65] (Figure 3).
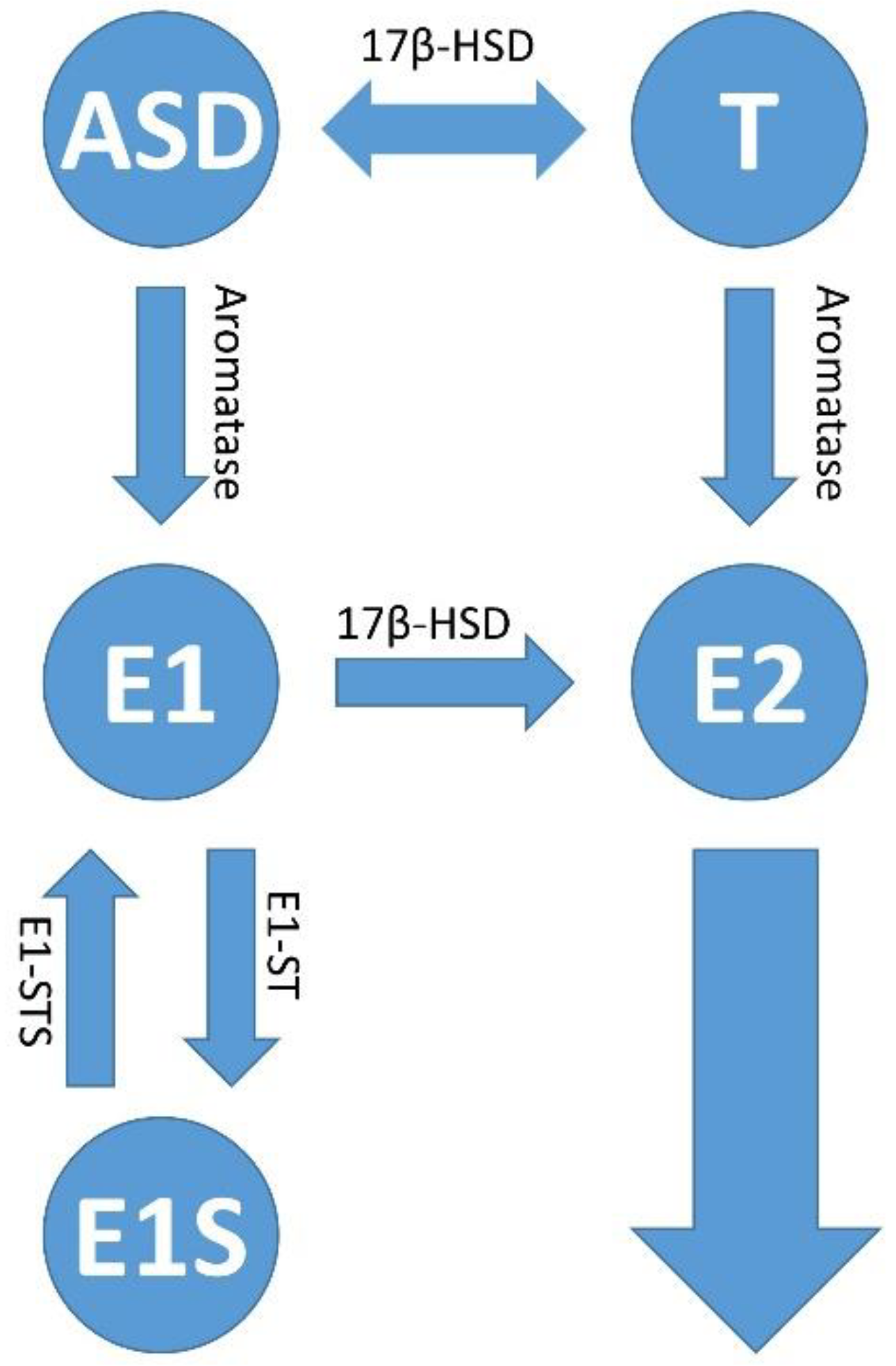
Figure 3. Estrogen synthesis pathways. In addition to estradiol (E2), estrone (E1) is also formed in the human body when androstenedione (ASD) is aromatized. This mainly happens in postmenopausal women. Estrone (E1) and/or estradiol (E2) are delivered to the tumor and its microenvironment via the bloodstream; or there is increased local production and retention of the hormones within the tumors. The latter occurs via increased activity of the cytochrome P450 enzyme, aromatase (CYP19A1), which converts androstenedione (ASD) to E1 and testosterone (T) to E2. The enzymatic activity of estrone (E1) sulfatase converts estrone sulfate (E1S) to estrone (E1); and 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1) activity is responsible for the conversion of E1 to E2, and androstenedione (ASD/AE) to testosterone (T), respectively. The sulfation of estradiol (E2) leads to the inactive form of estrogen, estrogen sulfate (E1S) via the activity of estrone sulfotransferase (E1-ST).
Estrone (E1) and estradiol (E2) are the starting molecules of estrogens, which are converted into metabolites in the liver and occur as hormones with different potency [66]. Subsequently, the starting molecules and their metabolites are converted to glucuronides and sulfates by glucuronidation or sulfation in order to be excreted via the bile, the kidneys or the intestines [67]. Just as estrogens are conjugated, they can also be de-conjugated by bacteria in the stomach and intestines. This occurs via β-glucuronidases (GUSB) or β-glucosidases from the class of glycosidases [68]. The bacteria in the intestine recycle the estradiol, and this can possibly fuel neoplasia, the development of which depends on estrogen signaling. The intestinal bacteria de-conjugate the bound estrogens and release them via the secretion of the GUSB so that they can carry out their biological activity in the body. When there is an oversupply of free estrogen via estrogen signaling, this can lead to hyper-estrogenic pathologies such as endometriosis and cancer. On the other hand, reduced GUSB activity caused by dysbiosis can lead to hypo-estrogenic pathologies such as obesity, metabolic syndrome and cardiovascular and neurological diseases [23][69]. This association between the β-glucuronidases (GUSB) produced by the gut microbiome, as part of the estrobolome that reactivates estrogens, has been confirmed [70]. Unconjugated estrogens are transferred back into the bloodstream via the intestines by means of GUSB enzymes and can develop their hormonal effect elsewhere.
In addition to the estrogen-metabolizing gut microbiome, there is also a microbiome in the gut, as mentioned earlier, that metabolizes tryptophan instead of estrogens. This microbial community, which also includes Klebsiella spp., does not compete with the host cells of the intestine for tryptophan and therefore promotes the production of melatonin. The gastric and intestinal cells and the endogenous metabolism in the liver produce melatonin as a secondary metabolite via pre-beta-lipoproteins (VLDL) and LDL [71]. A well-known effect of melatonin is that it regulates the circadian rhythm. In addition, it shows oncostatic effects such as the inhibition of proliferation and stimulation of apoptosis, immunomodulation and anti-inflammatory effects; furthermore, it shows supportive effects for the anti-oxidation systems. In addition, it regulates blood vessel formation [72]. The background to this is that ER signaling is blocked by melatonin, which is analogous to the effects of a selective estrogen receptor modulator (SERM) [73][74]. However, melatonin also shows the properties of a selective estrogen enzyme modulator (SEEM) [75]. Thus, melatonin inhibits the enzymatic activity of estrone (E1) sulfatase, which converts estrone sulfate (E1S) into estrone (E1), but also the activity of 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1), which is responsible for the conversion of E1 to E2. This subsequently reduces the circulating estradiol levels in the plasma [76]. Melatonin also promotes the sulfation of estradiol to the inactive form of estrogen, estrogen sulfate (E1S), via the activity of estrone sulfotransferase (E1-ST), and thus shows an anti-estrogenic effect. The effects of estradiol (E2) are diverse; they promote, in addition to growth, for example, the cervical epithelium, the immunosuppression in the microenvironment of the intraepithelial cervical lesions via their genomic effect on the estrogen receptor alpha (ERα). This also affects the cells responsible for immunosuppression such as the regulatory T cells (Tregs), the myeloid-derived suppressor cells (MDSCs) and cancer-associated fibroblasts (CAFs) [15].
2.2. Estrogen Signaling
Estradiol signals in a ligand-dependent and classical manner via the estrogen receptors α and β (ER α/β) and the G protein-coupled estrogen receptor 1 (GPER1), also known as G protein-coupled receptor 30 (GPR30). Independently, ligand signals in a non-classical way via other receptors such as the epidermal growth factor receptor (EGFR), the insulin growth factor receptor (IGFR) and the fibroblast growth factor receptor (FGFR) [77]. In addition, a genomic pathway is distinguished from a non-genomic pathway. In the genomic pathway, the hormone-receptor complexes are translocated to the nucleus, where the receptor dimers bind to estrogen-responsive elements (EREs) on the target gene promoter, triggering gene activation and epigenetic changes. The estrogen receptor serves as a transcription factor that regulates gene expression and thus cell proliferation and cell survival. Tissues that respond to fluctuating estrogen levels may experience organic changes [23]. As already mentioned in the last subchapter, one should not underestimate the indirect effects of estrogen signaling on the tumor microenvironment, such as the infiltrating immune cells, stroma cells and cancer-associated fibroblasts (CAFs) [77].
2.3. Influence of Estrogen Signaling on the Microenvironment of Cervical Intraepithelial Lesion, Cervical Pre-Cancer and in Tumor
On cervical carcinogenesis, animal models have made their decisive contribution to elucidating the mechanisms of the course of infection with the high-risk HPV types and the resulting transformation of the cervical lesions. In a frequently cited one by Arbeit et al. from 1996 [78], it was investigated the processes of carcinogenesis in a transgenic mouse model (K14-HPV16-E6/E7), which was permanently exposed to estradiol [78]. As already mentioned, chronic unresolved infection with one of the high-risk HPV strains is necessary for the start of the malignant transformation. However, further supporting co-factors, such as estradiol, are required for the full development of invasive carcinoma [79][80][81]. Sirtuin-1 (SIRT-1) cooperates with estrogen signaling in breast cancer tumorigenesis and progression [82]. SIRT-1 is also up-regulated in cervical carcinogenesis and leads via deacetylation to the destabilization of the tumor suppressor protein Werner syndrome protein (WRN). In HPV16 cervical cells, the destabilization of WRN resulted in increased basal cell proliferation, damage to DNA and epithelial thickening. This led to increased DNA replication via E1 and E2 and, as a result, to an increase in the proliferation of keratinocytes and the HPV life cycle in the lesions of the cervical neoplastic tissue [83]. It is also assumed that the likely use of birth control drugs and multiple maternities increases the risk of developing cervical squamous cell carcinoma (SCC) [84][85][86][87][88]. The association between viral infection and the effects of cofactors such as hormonal contraceptives in cervical carcinogenesis was examined by Marks MA et al. [89]. Analyzing cervical secretions from elderly women with HPV infection it was found that markers associated with anti-inflammation and allergies (IL-5, IL-9, IL-13, IL-17, EOTAXIN, GM-CSF and MIP-1α) were increased compared to HPV-negative women. In addition, T cell cytokines were shifted from interleukin 2 (IL-2) towards Eotaxin. This suggests immunosuppressive T cell responses via type 2 T helper cells (TH2 lymphocytes) [90]. Since estradiol, together with other factors, seems to suppress the immune response in cervical carcinogenesis, it is important to determine the estradiol concentrations in the blood plasma but also in the cervical secretions, namely the cervical mucus in postmenopausal HPV-infected women. During pregnancy, elevated levels of estradiol are found in blood plasma. Denmark found a higher rate of mortality with cervical cancer during pregnancy or shortly after pregnancy [91].
2.3.1. Estrogen Distribution in the Tumor and the Surrounding Microenvironment
Estrone (E1) and/or estradiol (E2) are delivered to the tumor and its microenvironment via the bloodstream or there is an increased local production and retention of the hormones within the tumors. The latter occurs via the increased activity of the cytochrome P450 enzyme, aromatase (CYP19A1), which converts androstenedione and testosterone into E2 [92]. Estradiol levels have been shown to increase in the tumor microenvironment (TME) of cervical carcinoma while present at regular levels in blood plasma. In the transformed cervical keratinocytes, estradiol was distributed within the cytoplasm and in the infiltrating immune cells, and in the stroma in both the cytoplasm and the nucleus [93][94]. Compared to estradiol levels, aromatase showed an analogous distribution, suggesting that the hormone is synthesized within the tumor microenvironment and tumor [93][95]. As an alternative to the synthesis pathways via aromatase, estradiol can also be synthesized in the tissue via other pathways. Estrone sulfate (E1S) is converted into estrone by means of estrone sulfatase (E1-STS) and then further into estradiol (E2) by means of the enzyme 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1). The two enzymes E1-STS and 17β-HSD were detected in vitro in the HPV-positive cervical carcinoma cell line Hela, but not in vivo or in xenografts from patients [65][96].
2.3.2. Different Estrogen Signaling Pathways Causing Both, Pro- and Anti-Tumorigenic Effects in HPV-Positive Lesions of the Cervical Epithelium
Depending on the respective concentration of estrogens in the tissue of the cervix, there is a proliferation of the cancer cells and the suppression of their apoptosis induction at physiological hormone levels [97][98][99][100]. On the other hand, an opposite effect can also set in with a higher distribution of estradiol in the tissue, which disrupts protein translation on the ribosomes and leads to apoptotic cancer cell death [101][102]. The latter pathway, at high hormone concentrations, via estradiol-induced phosphodiesterase 3A (PDE3A) activation and transactivation of Schlafen, a family member 12 (SLFN12) protein that is predicted to act upstream or within the negative regulation of cell proliferation, leads to the inhibition of mitochondrial activity and the blockade of the apoptotic proteins Bcl2 and Mcl1 for cancer cell apoptosis [103].
In addition to the need for E2, the transgenic mouse models K14-HPV16/HPV18-E6/E7 were also dependent on the expression of the estrogen receptor α (ERα) [78][104]. It was not surprising that selective estrogen receptor modulators (SERMs) and selective estrogen receptor disruptors (SERDs) inhibited the growth of precancerous lesions and carcinomas in animal models [78][80][105][106][107][108]. It was surprising to see that in the transformed cervical keratinocytes from healthy tissue in the direction of malignant development, the expression of ERα was gradually lost, and in mature cancer, no ERα expression could be detected, while, in contrast, the expression of ERβ was preserved and both ERα and ERβ expression were determined in the surrounding stroma [109][110][111][112][113][114]. If tumor cells cannot use estradiol via the genomic pathway, the estradiol originating from the tumor microenvironment can also signal in the tumor cell via non-genomic pathways [77]. The pro-tumor and anti-tumor effects of estradiol on malignant lesions due to HPV infection are not contradictory [13]. Since carcinogenesis always depends on the interactions between the developing malignancy and its tumor microenvironment [27][115], it must focus the attention primarily on the bidirectional crosstalk between the tumor and its tumor microenvironment. These primarily include the HPV-infected keratinocytes of the cervical lesion, but also the immune cells infiltrating the tumor stroma, which play a key role in deciding the fate of the lesion towards malignancy [108][116][117].It will discuss the hormonal effects on the tumor stroma and its effects on the infiltration of immune-modulating cells of the immune response.
2.3.3. The Importance of the ERα for the Tumor Stroma and the Tumor Microenvironment in Relation to the Development of Precancerous Lesions up to the Invasive Form of Cervical Carcinoma
Many benign cells of the cervical stroma express the estrogen receptor α. On the other hand, adequate estradiol levels are not produced in the blood plasma of women who are currently in their menstrual phase. This suggests that the hormone levels appropriate for ERα expression are produced by the hormone-sensitive tissues themselves [109][112][113]. The estrogen receptor α was expressed in 30% to more than 50% of the stromal cells surrounding the tumor from precancerous lesions and invasive cervical carcinoma, but not in the tumor itself. Immunohistochemistry showed inequality in the distribution of the receptor in the tumor stroma. It was present in all tumors, regardless of cancer grade, in the intracellular spaces between tumor cells in cancer-associated fibroblasts (CAFs), MDSCs and other subtypes of lymphocytes. The tumor cells themselves, on the other hand, did not show any ERα expression [93][94][113][118][119][120]. This relationship between the estrogen signaling of cervical squamous tumor cells and the surrounding stromal cells could only be explained by a paracrine delivery of estrogen signaling-derived signals from the stromal cells. In fact, a special procedure applied within the related transgenic mouse models helped to remove the ERα only from the stromal cells in order to demonstrate these paracrine mechanisms of estrogen signaling [110][121]. In addition to the results from the animal models, it was shown, in the human model of cervical carcinoma, that cancer-associated fibroblasts (CAFs), which were cultivated ex vivo, were estrogen receptor α positive. Thus, ERα genomic signaling in the CAFs induced the secretion of soluble molecules, which, in a paracrine manner, directly benefited malignant cell proliferation and migration, vascular angiogenesis and the epithelial–mesenchymal transition (EMT) of the metastatic tumor, and indirectly supported inflammatory processes within the cervical lesions that promoted cervical carcinogenesis [94]. Treating CAFs with a selective estrogen receptor modulator (SERM), in this case, methylpiperidinopyrazole (MPP), or a selective estrogen receptor disruptor (SERD), in this case, fulvestrant (Faslodex, ICI-182780), suppressed cell cycle- and metabolism-related genes in their expression. This attenuated tumorigenesis and angiogenesis as well as the functionality of estrogen signaling [94]. However, since the stromal genes, which are up-regulated via ERα signaling, are extremely important for cervical carcinogenesis [94][108][110][122], this implicates, conversely, that these genes should be therapeutically targeted [123].
2.3.4. Estrogen Signaling in the Infiltrating Cells of the Immune Response in Cervical Carcinogenesis
Since the cervical epithelium slowly loses its ERα expression in the course of carcinogenesis via the various stages of cervical intraepithelial neoplasia (CIN1–3) to invasive CxCa, estradiol (E2) likely initially acts via the classical genomic pathway, with decreasing ERα-Expression levels within the transformed cells possibly via the non-classical, non-genomic pathways. Furthermore, E2 promotes an anti-inflammatory and regulatory immune microenvironment, which contributes decisively to the success of cervical carcinogenesis [89] .
Erythrocytes, granulocytes, monocytes and thrombocytes are formed during the myeloid hematopoiesis that takes place in the bone marrow. Estrogens induce this process and also enhance the mobility of MDSCs and their inherent immunosuppressive character [124]. In pregnant women, it has been shown that the increased estradiol levels in the blood plasma caused by pregnancy were sufficient to stimulate myeloid hematopoiesis and increase the mobility of MDSCs from the site of origin, the bone marrow, towards the spleen and local tumors. The ERα signaling of granulocytic myeloid suppressor cells (GrMDSCs) contributed to potentiating the level of immunosuppression within the tumor. This resulted in the progression of cervical carcinoma tumor cells that were ERα-negative on their own [118]. Ex vivo and in an orthotopic animal model of cervical carcinoma, the estrogen receptor antagonist fulvestrant was shown to be able to abrogate the immunosuppressive function of the tumor-infiltrating GrMDSCs [118].
In addition to the MDSCs, the regulatory T cells (Tregs or suppressor T cells) also suppress the immune response by infiltrating the tumor. Estradiol causes the expansion of the Tregs and induces the FOXP3 gene promoter, which is mainly responsible for the regulation of Tregs in mice [125][126]. However, it has been demonstrated that estradiol occurs in the tumor, its stroma and the infiltrating immune cells in human CxCa [93][95]. In comparison to other cells of the immune system, the intracellular estradiol levels were highest in the circulating, intratumoral Tregs and the Tregs of the draining lymph nodes [93]. In addition to the induction of the FOXP3 gene promoter, the associated FOXP3 expression and the maintenance of the control function of the Tregs, the immunosuppressive TGF-β and IL-10 cytokines were also released via the cell contact of the immunosuppressive Tregs. This was all controlled via ERα signaling [93]. It was not surprising that eight possible estrogen-responsive elements (EREs) were found in the FOXP3 locus of Foxp3+ regulatory T cells (FOXP3+ Tregs). The complex of the estrogen receptor α and estradiol was able to bind the translated FOXP3 protein product in the Tregs in humans [93]. Next to this classic genomic pathway of estrogen signaling, it could also be shown that Tregs could also signal via non-genomic (fast) signaling via the G protein-coupled estrogen receptor 1 (GPER1, GPR30) or the membrane-bound ERα. This led, via protein kinase B (Akt/PKB) phosphorylation, to the activation of programmed cell death protein 1 (PD1) signaling and/or increased expression of perforin. Perforin, a cytolytic protein, is found in the granules of cytotoxic T cells and NK cells, but also in Tregs. After degranulation, the cell membrane of the target cell is perforated. A pore is formed (hence the name perforin). Granzyme B enters the target cell and subsequently causes apoptotic cell death [127]. Thus, Tregs encountering cytotoxic T cells or NK cells in the tumor microenvironment could use the same mechanism to induce their apoptotic death by granzyme B and perforin, thus suppressing the immune response. Granzyme B and perforin are thus important for both the clearance of the malignant transformed tissue and its progression in vivo, and help to determine whether the pro- or anti-tumorigenic forces of the immune microenvironment fight for dominance within the cervical intraepithelial lesions and the tumor and its microenvironment prevail [127][128][129][130][131][132].
In contrast, selective estrogen receptor disruptors (SERDs) such as ICI 182,780 (fulvestrant, 7α,17β-[9-[(4,4,5,5,5-pentafluoropentyl)sulfinyl]nonyl]estra-1,3,5(10)-triene-3,17-diol) or RU 58,668 (11β,17β)-11-[4-[[5-[(4,4,5,5,5-pentafluoropentyl)sulfonyl]pentyl]-oxy]phenylestra-1,3,5,(10)-triene-3,17-diol) were used as antagonists of estrogen signaling. This resulted in the abrogation of the effects of estradiol on tumor-infiltrating Treg cells (CD4+CD25highCD127low) and the concomitant destruction of ERα and suppression of FOXP3 expression to terminate the suppressive effects of Tregs on both CD8+ and CD4+(CD4+CD25int) effector T cell subsets [93][133].
Another important type of immune cell is the M2 tumor-associated macrophages (M2-TAMs). These likewise immunosuppressive, tumor-promoting cells of the innate immune system are stimulated, at least in breast, ovarian and lung carcinomas, via the effect of estradiol to migrate into the tumors and secrete VEGF. The latter leads to positive feedback that prompts M2-TAMs to infiltrate the tumors in even larger numbers [77]. The proteinase inhibitor 9 (PI9) expression is increased via estrogen signaling in the immune cells, which suppresses the secretion of granzyme B (GrB) both endo- and exogenously [134][135]. The expression levels of the granzyme gene family are also down-regulated by the interaction of the HR HPV16 E7 oncoprotein with estrogen signaling [136]. Granzyme B is also secreted by keratinocytes. This suggests that estrogen signaling inhibits granzyme B expression via a similar effect in HPV-infected keratinocytes of the cervical epithelium in collaboration with HPV oncoproteins [137]. As it was mentioned earlier, perforin and granzyme B promote apoptosis [127]. Granzyme B also promotes the degradation of collagen, an extremely important component of the extracellular matrix (ECM). This mechanism paves the way for the infiltration of the tumor microenvironment by cytotoxic T lymphocytes [138][139]. However, since granzyme B expression is now down-regulated via the collaboration of E2 with the HR HPV16 E7 oncoprotein, it saves the developing transformed keratinocyte from apoptotic cell death and protects it by preventing the invasion of effector T cells into the premalignant lesion prior to recognition and elimination by the immune system [135][140]. Chemokines such as CC chemokine ligand 2 (CCL2) and CCL5 promote tumor progression. Their expression is also induced by the action of estradiol, at least in breast cancer [141]. Both CD8+ cytotoxic effector T lymphocytes (CTLs), the NK cell mobilizing CD4+ T lymphocytes (T helper cells) and the regulatory T cells (Tregs) show estrogen receptor α (ERα) expression, but Tregs, compared to the other lymphocyte subgroups, show a higher ERα expression [93].
2.4. The Effect of ER Antagonists in Modulating the Immune Microenvironment of the Premalignant Cervical Lesion and CxCa
The use of selective estrogen receptor disruptors (SERDs, ER antagonists, ICIs) has shown to counteract the effects of estradiol on estrogen signaling and subsequently on the immune response cells infiltrating the tumor and its stroma, including cancer-associated fibroblasts (CAFs), MDSCs and regulatory (Th2) T lymphocytes (Tregs) in cervical carcinoma [80][93][94][118].
As already mentioned, estradiol (E2) acts both classically and ligand-dependently via the classical ERs and membrane-bound receptors such as GPER1 (GPR30), and non-classically and ligand-independently via other receptors such as EGFR, IGFR and FGFR. This is also dependent and independent of FOXP3 expression and dependent and independent of the programmed cell death 1 ligand 1 (PD-L1)/programmed cell death protein 1 (PD1) pathway [125][128][129][130][132]. When using ICIs such as fulvestrant, under the action of exogenously supplied E2, after some time there was a lifting of the suppression of ER signaling and a renewed secretion of cytokines, probably via signals transmitted via extranuclear activity pathways (RAS/MAPK and PI3K/AKT). This happened non-canonically, and further, for example., via transcription factors such as the activating protein-1 (AP-1) and the specific protein-1 (Sp1) [54][93][142][143][144]. Given the variety of pathways in which effects can be exerted via estrogen signaling, it might make more sense to tackle the problem of estrogen signaling inhibition “at the root”, which is hormone synthesis. In addition to therapy with ER antagonists, the use of a group of anti-estrogens such as aromatase inhibitors (AIs) could offer a more effective approach in the therapy of CxCa. For patients with breast cancer, it was found that continued therapy with aromatase inhibitors subsequently also reduced the incidence of malignant changes in the cervix. A 10-year incidence of high-grade cervical dysplasia (CIN2–3) was found in the group that has been screened regularly (HR = 0.49; 95% CI, 0.27 to 0.90; p = 0.0212), especially in women over 50 years of age (HR = 0.34; 95% CI, 0.14 to 0.80; p = 0.014). The protective effect of tamoxifen monotherapy against low-grade cervical dysplasia (CIN1) was found only in young women [145]. In another, the main load of CxCa (43%) was found in postmenopausal women who also showed a high E2 concentration in the tumor [93]. This reinforces the importance of inhibition of local synthesis of E2 by AIs in the affected tissue [145]. Letrozole and anastrozole, two drugs from the group of AIs, led to a reduction in Th2 cytokines and an increase in Th1 cytokines as well as a simultaneous reduction in forkhead box protein P3 (FOXP3+) regulatory T cells (Tregs) in animal experiments as well as in humans [146][147]. Thus, the inhibition of E2 synthesis via the action of the AIs led to the reactivation of the immune function of (Th1) cytotoxic T lymphocytes (CTLs) [128][148] and the natural killer (NK) cells [140] within the tumor microenvironment. AIs abrogate the function of aromatase only between the conversion of androstenedione to estrone (E1) and the conversion of testosterone to estradiol (E2). It is important to remember that, especially in postmenopausal women, E2 is also synthesized via the conversion of the predominant form of the hormone in these women from estrone sulfate (E1S) to E1 by estrone sulphatase and further by type 1 17β-hydroxysteroid dehydrogenase (17β-HSD, HSD17B) [15][65][149][150].
References
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; de Sanjosé, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers 2016, 2, 16086.
- De Vuyst, H.; Clifford, G.M.; Nascimento, M.C.; Madeleine, M.M.; Franceschi, S. Prevalence and type distribution of human papillomavirus in carcinoma and intraepithelial neoplasia of the vulva, vagina and anus: A meta-analysis. Int. J. Cancer 2009, 124, 1626–1636.
- Wittekindt, C.; Wagner, S.; Sharma, S.J.; Wurdemann, N.; Knuth, J.; Reder, H.; Klussmann, J.P. HPV—A different view on Head and Neck Cancer. Laryngorhinootologie 2018, 97, S48–S113.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Brianti, P.; De Flammineis, E.; Mercuri, S.R. Review of HPV-related diseases and cancers. New Microbiol. 2017, 40, 80–85.
- Chesson, H.W.; Dunne, E.F.; Hariri, S.; Markowitz, L.E. The estimated lifetime probability of acquiring human papillomavirus in the United States. Sex Transm. Dis. 2014, 41, 660–664.
- de Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 2017, 141, 664–670.
- Kang, S.D.; Chatterjee, S.; Alam, S.; Salzberg, A.C.; Milici, J.; van der Burg, S.H.; Meyers, C. Effect of Productive Human Papillomavirus 16 Infection on Global Gene Expression in Cervical Epithelium. J. Virol. 2018, 92, e01261-18.
- McBride, A.A.; Munger, K. Expert Views on HPV Infection. Viruses 2018, 10, 94.
- McLaughlin-Drubin, M.E.; Meyers, C. Evidence for the coexistence of two genital HPV types within the same host cell in vitro. Virology 2004, 321, 173–180.
- McLaughlin-Drubin, M.E.; Meyers, J.; Munger, K. Cancer associated human papillomaviruses. Curr. Opin. Virol. 2012, 2, 459–466.
- McLaughlin-Drubin, M.E.; Munger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208.
- James, C.D.; Morgan, I.M.; Bristol, M.L. The Relationship between Estrogen-Related Signaling and Human Papillomavirus Positive Cancers. Pathogens 2020, 9, 403.
- Marks, M.A.; Gravitt, P.E.; Burk, R.D.; Studentsov, Y.; Farzadegan, H.; Klein, S.L. Progesterone and 17beta-estradiol enhance regulatory responses to human papillomavirus type 16 virus-like particles in peripheral blood mononuclear cells from healthy women. Clin. Vaccine Immunol. 2010, 17, 609–617.
- Jayshree, R.S. The Immune Microenvironment in Human Papilloma Virus-Induced Cervical Lesions-Evidence for Estrogen as an Immunomodulator. Front. Cell. Infect. Microbiol. 2021, 11, 649815.
- Siegel, R.L.; Fedewa, S.A.; Miller, K.D.; Goding-Sauer, A.; Pinheiro, P.S.; Martinez-Tyson, D.; Jemal, A. Cancer statistics for Hispanics/Latinos, 2015. CA Cancer J. Clin. 2015, 65, 457–480.
- Viens, L.J.; Henley, S.J.; Watson, M.; Markowitz, L.E.; Thomas, C.C.; Thompson, T.D.; Razzaghi, H.; Saraiya, M. Human Papillomavirus-Associated Cancers—United States, 2008–2012. Morb. Mortal. Wkly. Rep. 2016, 65, 661–666.
- Hellberg, D. Sex steroids and cervical cancer. Anticancer Res. 2012, 32, 3045–3054.
- Li, B.; Zhang, L.; Zhao, J.; Tan, G.; Zhang, W.; Zhang, N.; Tian, J.; Qu, P. The value of cytokine levels in triage and risk prediction for women with persistent high-risk human papilloma virus infection of the cervix. Infect. Agents Cancer 2019, 14, 16.
- Nguyen, H.H.; Broker, T.R.; Chow, L.T.; Alvarez, R.D.; Vu, H.L.; Andrasi, J.; Brewer, L.R.; Jin, G.; Mestecky, J. Immune responses to human papillomavirus in genital tract of women with cervical cancer. Gynecol. Oncol. 2005, 96, 452–461.
- Samir, R.; Asplund, A.; Tot, T.; Pekar, G.; Hellberg, D. High-risk HPV infection and CIN grade correlates to the expression of c-myc, CD4+, FHIT, E-cadherin, Ki-67, and p16INK4a. J. Low. Genit. Tract Dis. 2011, 15, 280–286.
- Silins, I.; Kallings, I.; Dillner, J. Correlates of the spread of human papillomavirus infection. Cancer Epidemiol. Biomarkers Prev. 2000, 9, 953–959.
- Baker, J.M.; Al-Nakkash, L.; Herbst-Kralovetz, M.M. Estrogen-gut microbiome axis: Physiological and clinical implications. Maturitas 2017, 103, 45–53.
- Ratten, L.K.; Plummer, E.L.; Bradshaw, C.S.; Fairley, C.K.; Murray, G.L.; Garland, S.M.; Bateson, D.; Tachedjian, G.; Masson, L.; Vodstrcil, L.A. The Effect of Exogenous Sex Steroids on the Vaginal Microbiota: A Systematic Review. Front. Cell. Infect. Microbiol. 2021, 11, 732423.
- Zhou, Z.W.; Long, H.Z.; Cheng, Y.; Luo, H.Y.; Wen, D.D.; Gao, L.C. From Microbiome to Inflammation: The Key Drivers of Cervical Cancer. Front. Microbiol. 2021, 12, 767931.
- Wang, L.H.; Chen, L.R.; Chen, K.H. In Vitro and Vivo Identification, Metabolism and Action of Xenoestrogens: An Overview. Int. J. Mol. Sci. 2021, 22, 4013.
- Läsche, M.; Urban, H.; Gallwas, J.; Gründker, C. HPV and Other Microbiota; Who’s Good and Who’s Bad: Effects of the Microbial Environment on the Development of Cervical Cancer-A Non-Systematic Review. Cells 2021, 10, 714.
- Jia, M.; Dahlman-Wright, K.; Gustafsson, J. Estrogen receptor alpha and beta in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 557–568.
- Taneja, V. Sex Hormones Determine Immune Response. Front. Immunol. 2018, 9, 1931.
- Amador-Molina, A.; Hernández-Valencia, J.F.; Lamoyi, E.; Contreras-Paredes, A.; Lizano, M. Role of innate immunity against human papillomavirus (HPV) infections and effect of adjuvants in promoting specific immune response. Viruses 2013, 5, 2624–2642.
- Evans, M.R.; James, C.D.; Bristol, M.L.; Nulton, T.J.; Wang, X.; Kaur, N.; White, E.A.; Windle, B.; Morgan, I.M. Human Papillomavirus 16 E2 Regulates Keratinocyte Gene Expression Relevant to Cancer and the Viral Life Cycle. J. Virol. 2019, 93, e01941-18.
- Woodworth, C.D. HPV innate immunity. Front. Biosci. 2002, 7, d2058–d2071.
- Patel, S.; Homaei, A.; Raju, A.B.; Meher, B.R. Estrogen: The necessary evil for human health, and ways to tame it. Biomed. Pharmacother. 2018, 102, 403–411.
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 2017, 13, e1006211.
- Ferber, M.J.; Thorland, E.C.; Brink, A.A.; Rapp, A.K.; Phillips, L.A.; McGovern, R.; Gostout, B.S.; Cheung, T.H.; Chung, T.K.; Fu, W.Y.; et al. Preferential integration of human papillomavirus type 18 near the c-myc locus in cervical carcinoma. Oncogene 2003, 22, 7233–7242.
- Peter, M.; Rosty, C.; Couturier, J.; Radvanyi, F.; Teshima, H.; Sastre-Garau, X. MYC activation associated with the integration of HPV DNA at the MYC locus in genital tumors. Oncogene 2006, 25, 5985–5993.
- Smith, P.P.; Friedman, C.L.; Bryant, E.M.; McDougall, J.K. Viral integration and fragile sites in human papillomavirus-immortalized human keratinocyte cell lines. Genes Chromosomes Cancer 1992, 5, 150–157.
- Arias-Pulido, H.; Peyton, C.L.; Joste, N.E.; Vargas, H.; Wheeler, C.M. Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. J. Clin. Microbiol. 2006, 44, 1755–1762.
- Cannarella, R.; Condorelli, R.A.; Mongioì, L.M.; Barbagallo, F.; Calogero, A.E.; La Vignera, S. Effects of the selective estrogen receptor modulators for the treatment of male infertility: A systematic review and meta-analysis. Expert Opin. Pharmacother. 2019, 20, 1517–1525.
- Cauley, J.A. Estrogen and bone health in men and women. Steroids 2015, 99, 11–15.
- Dobbs, R.W.; Malhotra, N.R.; Greenwald, D.T.; Wang, A.Y.; Prins, G.S.; Abern, M.R. Estrogens and prostate cancer. Prostate Cancer Prostatic Dis. 2019, 22, 185–194.
- Harrison, R.F.; Bonnar, J. Clinical uses of estrogens. Pharmacol. Ther. 1980, 11, 451–467.
- Hasegawa, Y.; Itonaga, T.; Ikegawa, K.; Nishigaki, S.; Kawai, M.; Koga, E.; Sakakibara, H.; Ross, J.L. Ultra-low-dose estrogen therapy for female hypogonadism. Clin. Pediatr. Endocrinol. 2020, 29, 49–53.
- Knowlton, A.A.; Lee, A.R. Estrogen and the cardiovascular system. Pharmacol. Ther. 2012, 135, 54–70.
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell. Immunol. 2015, 294, 63–69.
- Lu, C.L.; Herndon, C. New roles for neuronal estrogen receptors. Neurogastroenterol. Motil. 2017, 29, e13121.
- Marquardt, R.M.; Kim, T.H.; Shin, J.H.; Jeong, J.W. Progesterone and Estrogen Signaling in the Endometrium: What Goes Wrong in Endometriosis? Int. J. Mol. Sci. 2019, 20, 3822.
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The role of estrogens in control of energy balance and glucose homeostasis. Endocr. Rev. 2013, 34, 309–338.
- Nakano, T.; Kadono, Y.; Iwamoto, H.; Yaegashi, H.; Iijima, M.; Kawaguchi, S.; Nohara, T.; Shigehara, K.; Izumi, K.; Mizokami, A. Therapeutic Effect of Ethinylestradiol in Castration-resistant Prostate Cancer. Anticancer Res. 2020, 40, 2291–2296.
- Pett, M.; Coleman, N. Integration of high-risk human papillomavirus: A key event in cervical carcinogenesis? J. Pathol. 2007, 212, 356–367.
- Regidor, P.A. Clinical relevance in present day hormonal contraception. Horm. Mol. Biol. Clin. Investig. 2018, 37.
- Simpson, E.R. Sources of estrogen and their importance. J. Steroid Biochem. Mol. Biol. 2003, 86, 225–230.
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170.
- Hariri, L.; Rehman, A. Estradiol. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022.
- Caroccia, B.; Seccia, T.M.; Barton, M.; Rossi, G.P. Estrogen Signaling in the Adrenal Cortex: Implications for Blood Pressure Sex Differences. Hypertension 2016, 68, 840–848.
- Nelson, L.R.; Bulun, S.E. Estrogen production and action. J. Am. Acad. Dermatol. 2001, 45, S116–S124.
- Wasada, T.; Akamine, Y.; Kato, K.; Ibayashi, H.; Nomura, Y. Adrenal contribution to circulating estrogens in woman. Endocrinol. Jpn 1978, 25, 123–128.
- Barakat, R.; Oakley, O.; Kim, H.; Jin, J.; Ko, C.J. Extra-gonadal sites of estrogen biosynthesis and function. BMB Rep. 2016, 49, 488–496.
- Cui, J.; Shen, Y.; Li, R. Estrogen synthesis and signaling pathways during aging: From periphery to brain. Trends Mol. Med. 2013, 19, 197–209.
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400.
- Marchesi, J.R.; Ravel, J. The vocabulary of microbiome research: A proposal. Microbiome 2015, 3, 31.
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533.
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359.
- Plottel, C.S.; Blaser, M.J. Microbiome and malignancy. Cell Host Microbe 2011, 10, 324–335.
- Gruber, C.J.; Tschugguel, W.; Schneeberger, C.; Huber, J.C. Production and actions of estrogens. N. Engl. J. Med. 2002, 346, 340–352.
- Zhu, B.T.; Han, G.Z.; Shim, J.Y.; Wen, Y.; Jiang, X.R. Quantitative structure-activity relationship of various endogenous estrogen metabolites for human estrogen receptor alpha and beta subtypes: Insights into the structural determinants favoring a differential subtype binding. Endocrinology 2006, 147, 4132–4150.
- Raftogianis, R.; Creveling, C.; Weinshilboum, R.; Weisz, J. Estrogen metabolism by conjugation. J. Natl. Cancer Inst. Monogr. 2000, 2000, 113–124.
- Dabek, M.; McCrae, S.I.; Stevens, V.J.; Duncan, S.H.; Louis, P. Distribution of beta-glucosidase and beta-glucuronidase activity and of beta-glucuronidase gene gus in human colonic bacteria. FEMS Microbiol. Ecol. 2008, 66, 487–495.
- Chen, K.L.; Madak-Erdogan, Z. Estrogen and Microbiota Crosstalk: Should We Pay Attention? Trends Endocrinol. Metab. 2016, 27, 752–755.
- Ervin, S.M.; Li, H.; Lim, L.; Roberts, L.R.; Liang, X.; Mani, S.; Redinbo, M.R. Gut microbial β-glucuronidases reactivate estrogens as components of the estrobolome that reactivate estrogens. J. Biol. Chem. 2019, 294, 18586–18599.
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front Cell Infect. Microbiol. 2018, 8, 13.
- Talib, W.H. Melatonin and Cancer Hallmarks. Molecules 2018, 23, 518.
- Blask, D.E.; Hill, S.M. Effects of melatonin on cancer: Studies on MCF-7 human breast cancer cells in culture. J. Neural Transm. Suppl. 1986, 21, 433–449.
- Hill, S.M.; Blask, D.E. Effects of the pineal hormone melatonin on the proliferation and morphological characteristics of human breast cancer cells (MCF-7) in culture. Cancer Res. 1988, 48, 6121–6126.
- Gonzalez, A.; Cos, S.; Martinez-Campa, C.; Alonso-Gonzalez, C.; Sanchez-Mateos, S.; Mediavilla, M.D.; Sanchez-Barcelo, E.J. Selective estrogen enzyme modulator actions of melatonin in human breast cancer cells. J. Pineal Res. 2008, 45, 86–92.
- Martínez-Campa, C.; González, A.; Mediavilla, M.D.; Alonso-González, C.; Alvarez-García, V.; Sánchez-Barceló, E.J.; Cos, S. Melatonin inhibits aromatase promoter expression by regulating cyclooxygenases expression and activity in breast cancer cells. Br. J. Cancer 2009, 101, 1613–1619.
- Somasundaram, A.; Rothenberger, N.J.; Stabile, L.P. The Impact of Estrogen in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1277, 33–52.
- Arbeit, J.M.; Howley, P.M.; Hanahan, D. Chronic estrogen-induced cervical and vaginal squamous carcinogenesis in human papillomavirus type 16 transgenic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 2930–2935.
- Brake, T.; Lambert, P.F. Estrogen contributes to the onset, persistence, and malignant progression of cervical cancer in a human papillomavirus-transgenic mouse model. Proc. Natl. Acad. Sci. USA 2005, 102, 2490–2495.
- Chung, S.H.; Lambert, P.F. Prevention and treatment of cervical cancer in mice using estrogen receptor antagonists. Proc. Natl. Acad. Sci. USA 2009, 106, 19467–19472.
- Muñoz, N.; Castellsagué, X.; Berrington de González, A.; Gissmann, L. Chapter 1: HPV in the etiology of human cancer. Vaccine 2006, 24 (Suppl. 3), S1–S10.
- Liarte, S.; Alonso-Romero, J.L.; Nicolás, F.J. SIRT1 and Estrogen Signaling Cooperation for Breast Cancer Onset and Progression. Front. Endocrinol. 2018, 9, 552.
- James, C.D.; Das, D.; Morgan, E.L.; Otoa, R.; Macdonald, A.; Morgan, I.M. Werner Syndrome Protein (WRN) Regulates Cell Proliferation and the Human Papillomavirus 16 Life Cycle during Epithelial Differentiation. mSphere 2020, 5, e00858-20.
- Jensen, K.E.; Schmiedel, S.; Norrild, B.; Frederiksen, K.; Iftner, T.; Kjaer, S.K. Parity as a cofactor for high-grade cervical disease among women with persistent human papillomavirus infection: A 13-year follow-up. Br. J. Cancer 2013, 108, 234–239.
- Moreno, V.; Bosch, F.X.; Muñoz, N.; Meijer, C.J.; Shah, K.V.; Walboomers, J.M.; Herrero, R.; Franceschi, S. Effect of oral contraceptives on risk of cervical cancer in women with human papillomavirus infection: The IARC multicentric case-control study. Lancet 2002, 359, 1085–1092.
- Muñoz, N.; Franceschi, S.; Bosetti, C.; Moreno, V.; Herrero, R.; Smith, J.S.; Shah, K.V.; Meijer, C.J.; Bosch, F.X. Role of parity and human papillomavirus in cervical cancer: The IARC multicentric case-control study. Lancet 2002, 359, 1093–1101.
- Rinaldi, S.; Plummer, M.; Biessy, C.; Castellsagué, X.; Overvad, K.; Krüger Kjær, S.; Tjønneland, A.; Clavel-Chapelon, F.; Chabbert-Buffet, N.; Mesrine, S.; et al. Endogenous sex steroids and risk of cervical carcinoma: Results from the EPIC study. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 2532–2540.
- Roura, E.; Travier, N.; Waterboer, T.; de Sanjosé, S.; Bosch, F.X.; Pawlita, M.; Pala, V.; Weiderpass, E.; Margall, N.; Dillner, J.; et al. The Influence of Hormonal Factors on the Risk of Developing Cervical Cancer and Pre-Cancer: Results from the EPIC Cohort. PLoS ONE 2016, 11, e0147029.
- Marks, M.; Klein, S.; Gravitt, P.; Feinstone, H. Hormonal contraception and HPV: A tale of differing and overlapping mechanisms. Open Access J. Contracept. 2011, 2, 161–174.
- Marks, M.A.; Viscidi, R.P.; Chang, K.; Silver, M.; Burke, A.; Howard, R.; Gravitt, P.E. Differences in the concentration and correlation of cervical immune markers among HPV positive and negative perimenopausal women. Cytokine 2011, 56, 798–803.
- Eibye, S.; Krüger Kjær, S.; Nielsen, T.S.; Mellemkjær, L. Mortality Among Women With Cervical Cancer During or Shortly After a Pregnancy in Denmark 1968 to 2006. Int. J. Gynecol. Cancer 2016, 26, 951–958.
- Lønning, P.E.; Haynes, B.P.; Straume, A.H.; Dunbier, A.; Helle, H.; Knappskog, S.; Dowsett, M. Exploring breast cancer estrogen disposition: The basis for endocrine manipulation. Clin. Cancer Res. 2011, 17, 4948–4958.
- Adurthi, S.; Kumar, M.M.; Vinodkumar, H.S.; Mukherjee, G.; Krishnamurthy, H.; Acharya, K.K.; Bafna, U.D.; Uma, D.K.; Abhishekh, B.; Krishna, S.; et al. Oestrogen Receptor-α binds the FOXP3 promoter and modulates regulatory T-cell function in human cervical cancer. Sci. Rep. 2017, 7, 17289.
- Kumar, M.M.; Davuluri, S.; Poojar, S.; Mukherjee, G.; Bajpai, A.K.; Bafna, U.D.; Devi, U.K.; Kallur, P.P.; Kshitish, A.K.; Jayshree, R.S. Role of estrogen receptor alpha in human cervical cancer-associated fibroblasts: A transcriptomic study. Tumor Biol. 2016, 37, 4409–4420.
- Nair, H.B.; Luthra, R.; Kirma, N.; Liu, Y.G.; Flowers, L.; Evans, D.; Tekmal, R.R. Induction of aromatase expression in cervical carcinomas: Effects of endogenous estrogen on cervical cancer cell proliferation. Cancer Res. 2005, 65, 11164–11173.
- Fournier, M.A.; Poirier, D. Estrogen formation in endometrial and cervix cancer cell lines: Involvement of aromatase, steroid sulfatase and 17beta-hydroxysteroid dehydrogenases (types 1, 5, 7 and 12). Mol. Cell. Endocrinol. 2009, 301, 142–145.
- Chen, Y.H.; Huang, L.H.; Chen, T.M. Differential effects of progestins and estrogens on long control regions of human papillomavirus types 16 and 18. Biochem. Biophys. Res. Commun. 1996, 224, 651–659.
- Kim, C.J.; Um, S.J.; Kim, T.Y.; Kim, E.J.; Park, T.C.; Kim, S.J.; Namkoong, S.E.; Park, J.S. Regulation of cell growth and HPV genes by exogenous estrogen in cervical cancer cells. Int. J. Gynecol. Cancer 2000, 10, 157–164.
- Mitrani-Rosenbaum, S.; Tsvieli, R.; Tur-Kaspa, R. Oestrogen stimulates differential transcription of human papillomavirus type 16 in SiHa cervical carcinoma cells. J. Gen. Virol. 1989, 70 Pt 8, 2227–2232.
- Ruutu, M.; Wahlroos, N.; Syrjänen, K.; Johansson, B.; Syrjänen, S. Effects of 17beta-estradiol and progesterone on transcription of human papillomavirus 16 E6/E7 oncogenes in CaSki and SiHa cell lines. Int. J. Gynecol. Cancer 2006, 16, 1261–1268.
- Bristol, M.L.; James, C.D.; Wang, X.; Fontan, C.T.; Morgan, I.M. Estrogen Attenuates the Growth of Human Papillomavirus-Positive Epithelial Cells. mSphere 2020, 5, e00049-20.
- Li, D.; Chen, J.; Ai, Y.; Gu, X.; Li, L.; Che, D.; Jiang, Z.; Chen, S.; Huang, H.; Wang, J.; et al. Estrogen-Related Hormones Induce Apoptosis by Stabilizing Schlafen-12 Protein Turnover. Mol. Cell 2019, 75, 1103–1116.e9.
- Lamb, H.M.; Hardwick, J.M. The Dark Side of Estrogen Stops Translation to Induce Apoptosis. Mol. Cell 2019, 75, 1087–1089.
- Park, J.S.; Rhyu, J.W.; Kim, C.J.; Kim, H.S.; Lee, S.Y.; Kwon, Y.I.; Namkoong, S.E.; Sin, H.S.; Um, S.J. Neoplastic change of squamo-columnar junction in uterine cervix and vaginal epithelium by exogenous estrogen in hpv-18 URR E6/E7 transgenic mice. Gynecol. Oncol. 2003, 89, 360–368.
- Chung, S.H.; Franceschi, S.; Lambert, P.F. Estrogen and ERalpha: Culprits in cervical cancer? Trends Endocrinol. Metab. 2010, 21, 504–511.
- Chung, S.H.; Wiedmeyer, K.; Shai, A.; Korach, K.S.; Lambert, P.F. Requirement for estrogen receptor alpha in a mouse model for human papillomavirus-associated cervical cancer. Cancer Res. 2008, 68, 9928–9934.
- Riley, R.R.; Duensing, S.; Brake, T.; Münger, K.; Lambert, P.F.; Arbeit, J.M. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003, 63, 4862–4871.
- Spurgeon, M.E.; den Boon, J.A.; Horswill, M.; Barthakur, S.; Forouzan, O.; Rader, J.S.; Beebe, D.J.; Roopra, A.; Ahlquist, P.; Lambert, P.F. Human papillomavirus oncogenes reprogram the cervical cancer microenvironment independently of and synergistically with estrogen. Proc. Natl. Acad. Sci. USA 2017, 114, E9076–E9085.
- Coelho, F.R.; Prado, J.C.; Pereira Sobrinho, J.S.; Hamada, G.; Landman, G.; Pinto, C.A.; Nonogaki, S.; Villa, L.L. Estrogen and progesterone receptors in human papilloma virus-related cervical neoplasia. Braz. J. Med. Biol. Res. 2004, 37, 83–88.
- den Boon, J.A.; Pyeon, D.; Wang, S.S.; Horswill, M.; Schiffman, M.; Sherman, M.; Zuna, R.E.; Wang, Z.; Hewitt, S.M.; Pearson, R.; et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E3255–E3264.
- Konishi, I.; Fujii, S.; Nonogaki, H.; Nanbu, Y.; Iwai, T.; Mori, T. Immunohistochemical analysis of estrogen receptors, progesterone receptors, Ki-67 antigen, and human papillomavirus DNA in normal and neoplastic epithelium of the uterine cervix. Cancer 1991, 68, 1340–1350.
- López-Romero, R.; Garrido-Guerrero, E.; Rangel-López, A.; Manuel-Apolinar, L.; Piña-Sánchez, P.; Lazos-Ochoa, M.; Mantilla-Morales, A.; Bandala, C.; Salcedo, M. The cervical malignant cells display a down regulation of ER-α but retain the ER-β expression. Int. J. Clin. Exp. Pathol. 2013, 6, 1594–1602.
- Nonogaki, H.; Fujii, S.; Konishi, I.; Nanbu, Y.; Ozaki, S.; Ishikawa, Y.; Mori, T. Estrogen receptor localization in normal and neoplastic epithelium of the uterine cervix. Cancer 1990, 66, 2620–2627.
- Zhai, Y.; Bommer, G.T.; Feng, Y.; Wiese, A.B.; Fearon, E.R.; Cho, K.R. Loss of estrogen receptor 1 enhances cervical cancer invasion. Am. J. Pathol. 2010, 177, 884–895.
- Läsche, M.; Emons, G.; Gründker, C. Shedding New Light on Cancer Metabolism: A Metabolic Tightrope Between Life and Death. Front. Oncol. 2020, 10, 409.
- De Nola, R.; Menga, A.; Castegna, A.; Loizzi, V.; Ranieri, G.; Cicinelli, E.; Cormio, G. The Crowded Crosstalk between Cancer Cells and Stromal Microenvironment in Gynecological Malignancies: Biological Pathways and Therapeutic Implication. Int. J. Mol. Sci. 2019, 20, 2401.
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437.
- Kozasa, K.; Mabuchi, S.; Matsumoto, Y.; Kuroda, H.; Yokoi, E.; Komura, N.; Kawano, M.; Takahashi, R.; Sasano, T.; Shimura, K.; et al. Estrogen stimulates female cancer progression by inducing myeloid-derived suppressive cells: Investigations on pregnant and non-pregnant experimental models. Oncotarget 2019, 10, 1887–1902.
- Kwasniewska, A.; Postawski, K.; Gozdzicka-Jozefiak, A.; Kwasniewski, W.; Grywalska, E.; Zdunek, M.; Korobowicz, E. Estrogen and progesterone receptor expression in HPV-positive and HPV-negative cervical carcinomas. Oncol. Rep. 2011, 26, 153–160.
- Mosny, D.S.; Herholz, J.; Degen, W.; Bender, H.G. Immunohistochemical investigations of steroid receptors in normal and neoplastic squamous epithelium of the uterine cervix. Gynecol. Oncol. 1989, 35, 373–377.
- Chung, S.H.; Shin, M.K.; Korach, K.S.; Lambert, P.F. Requirement for stromal estrogen receptor alpha in cervical neoplasia. Horm. Cancer 2013, 4, 50–59.
- Spurgeon, M.E.; Lambert, P.F. Human Papillomavirus and the Stroma: Bidirectional Crosstalk during the Virus Life Cycle and Carcinogenesis. Viruses 2017, 9, 219.
- Son, J.; Park, Y.; Chung, S.H. Epithelial oestrogen receptor α is dispensable for the development of oestrogen-induced cervical neoplastic diseases. J. Pathol. 2018, 245, 147–152.
- Svoronos, N.; Perales-Puchalt, A.; Allegrezza, M.J.; Rutkowski, M.R.; Payne, K.K.; Tesone, A.J.; Nguyen, J.M.; Curiel, T.J.; Cadungog, M.G.; Singhal, S.; et al. Tumor Cell-Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov. 2017, 7, 72–85.
- Polanczyk, M.J.; Carson, B.D.; Subramanian, S.; Afentoulis, M.; Vandenbark, A.A.; Ziegler, S.F.; Offner, H. Cutting edge: Estrogen drives expansion of the CD4+CD25+ regulatory T cell compartment. J. Immunol. 2004, 173, 2227–2230.
- Yang, X.F. Factors regulating apoptosis and homeostasis of CD4+ CD25(high) FOXP3+ regulatory T cells are new therapeutic targets. Front. Biosci. 2008, 13, 1472–1499.
- Cao, X.; Cai, S.F.; Fehniger, T.A.; Song, J.; Collins, L.I.; Piwnica-Worms, D.R.; Ley, T.J. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 2007, 27, 635–646.
- Polanczyk, M.J.; Hopke, C.; Vandenbark, A.A.; Offner, H. Estrogen-mediated immunomodulation involves reduced activation of effector T cells, potentiation of Treg cells, and enhanced expression of the PD-1 costimulatory pathway. J. Neurosci. Res. 2006, 84, 370–378.
- Polanczyk, M.J.; Hopke, C.; Vandenbark, A.A.; Offner, H. Treg suppressive activity involves estrogen-dependent expression of programmed death-1 (PD-1). Int. Immunol. 2007, 19, 337–343.
- Prieto, G.A.; Rosenstein, Y. Oestradiol potentiates the suppressive function of human CD4 CD25 regulatory T cells by promoting their proliferation. Immunology 2006, 118, 58–65.
- Valor, L.; Teijeiro, R.; Aristimuño, C.; Faure, F.; Alonso, B.; de Andrés, C.; Tejera, M.; López-Lazareno, N.; Fernández-Cruz, E.; Sánchez-Ramón, S. Estradiol-dependent perforin expression by human regulatory T-cells. Eur. J. Clin. Investig. 2011, 41, 357–364.
- Yates, M.A.; Li, Y.; Chlebeck, P.J.; Offner, H. GPR30, but not estrogen receptor-alpha, is crucial in the treatment of experimental autoimmune encephalomyelitis by oral ethinyl estradiol. BMC Immunol. 2010, 11, 20.
- Traboulsi, T.; El Ezzy, M.; Gleason, J.L.; Mader, S. Antiestrogens: Structure-activity relationships and use in breast cancer treatment. J. Mol. Endocrinol. 2017, 58, R15–R31.
- Khan, M.S.; Singh, P.; Azhar, A.; Naseem, A.; Rashid, Q.; Kabir, M.A.; Jairajpuri, M.A. Serpin Inhibition Mechanism: A Delicate Balance between Native Metastable State and Polymerization. J. Amino Acids 2011, 2011, 606797.
- Medema, J.P.; de Jong, J.; Peltenburg, L.T.; Verdegaal, E.M.; Gorter, A.; Bres, S.A.; Franken, K.L.; Hahne, M.; Albar, J.P.; Melief, C.J.; et al. Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 11515–11520.
- Munguía-Moreno, J.A.; Díaz-Chavéz, J.; García-Villa, E.; Albino-Sanchez, M.E.; Mendoza-Villanueva, D.; Ocadiz-Delgado, R.; Bonilla-Delgado, J.; Marín-Flores, A.; Cortés-Malagón, E.M.; Alvarez-Rios, E.; et al. Early synergistic interactions between the HPV16-E7 oncoprotein and 17β-oestradiol for repressing the expression of Granzyme B in a cervical cancer model. Int. J. Oncol. 2018, 53, 579–591.
- Hernandez-Pigeon, H.; Jean, C.; Charruyer, A.; Haure, M.J.; Baudouin, C.; Charveron, M.; Quillet-Mary, A.; Laurent, G. UVA induces granzyme B in human keratinocytes through MIF: Implication in extracellular matrix remodeling. J. Biol. Chem. 2007, 282, 8157–8164.
- Parkinson, L.G.; Toro, A.; Zhao, H.; Brown, K.; Tebbutt, S.J.; Granville, D.J. Granzyme B mediates both direct and indirect cleavage of extracellular matrix in skin after chronic low-dose ultraviolet light irradiation. Aging Cell 2015, 14, 67–77.
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910.
- Jiang, X.; Orr, B.A.; Kranz, D.M.; Shapiro, D.J. Estrogen induction of the granzyme B inhibitor, proteinase inhibitor 9, protects cells against apoptosis mediated by cytotoxic T lymphocytes and natural killer cells. Endocrinology 2006, 147, 1419–1426.
- Svensson, S.; Abrahamsson, A.; Rodriguez, G.V.; Olsson, A.K.; Jensen, L.; Cao, Y.; Dabrosin, C. CCL2 and CCL5 Are Novel Therapeutic Targets for Estrogen-Dependent Breast Cancer. Clin. Cancer Res. 2015, 21, 3794–3805.
- deGraffenried, L.A.; Hilsenbeck, S.G.; Fuqua, S.A. Sp1 is essential for estrogen receptor alpha gene transcription. J. Steroid Biochem. Mol. Biol. 2002, 82, 7–18.
- Marino, M.; Galluzzo, P.; Ascenzi, P. Estrogen signaling multiple pathways to impact gene transcription. Curr. Genom. 2006, 7, 497–508.
- Webb, P.; Nguyen, P.; Valentine, C.; Lopez, G.N.; Kwok, G.R.; McInerney, E.; Katzenellenbogen, B.S.; Enmark, E.; Gustafsson, J.A.; Nilsson, S.; et al. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol. Endocrinol. 1999, 13, 1672–1685.
- Hsieh, C.J.; Hong, M.K.; Chen, P.C.; Wang, J.H.; Chu, T.Y. Antiestrogen use reduces risk of cervical neoplasia in breast cancer patients: A population-based study. Oncotarget 2017, 8, 29361–29369.
- Generali, D.; Bates, G.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Bersiga, A.; Allevi, G.; Milani, M.; Aguggini, S.; et al. Immunomodulation of FOXP3+ regulatory T cells by the aromatase inhibitor letrozole in breast cancer patients. Clin. Cancer Res. 2009, 15, 1046–1051.
- Ray, A.; Ficek, M. Immunomodulatory effects of anti-estrogenic drugs. Acta Pharm. 2012, 62, 141–155.
- Polese, B.; Gridelet, V.; Araklioti, E.; Martens, H.; Perrier d’Hauterive, S.; Geenen, V. The Endocrine Milieu and CD4 T-Lymphocyte Polarization during Pregnancy. Front. Endocrinol. 2014, 5, 106.
- Purohit, A.; Foster, P.A. Steroid sulfatase inhibitors for estrogen- and androgen-dependent cancers. J. Endocrinol. 2012, 212, 99–110.
- Secky, L.; Svoboda, M.; Klameth, L.; Bajna, E.; Hamilton, G.; Zeillinger, R.; Jäger, W.; Thalhammer, T. The sulfatase pathway for estrogen formation: Targets for the treatment and diagnosis of hormone-associated tumors. J. Drug Deliv. 2013, 2013, 957605.
More
Information
Subjects:
Endocrinology & Metabolism
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
2 times
(View History)
Update Date:
16 May 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No