The special thing about an HPV infection is that it is not only able to trick the immune system in a sophisticated way, but also, through genetic integration into the host genome, to use all the resources available to the host cells to complete the replication cycle of the virus without activating the alarm mechanisms of immune recognition and elimination. The mechanisms utilized by the virus are the metabolic, immune, and hormonal signaling pathways that it manipulates. Since the virus is dependent on replication enzymes of the host cells, it also intervenes in the cell cycle of the differentiating keratinocytes and shifts their terminal differentiation to the uppermost layers of the squamocolumnar transformation zone (TZ) of the cervix.
- cervical cancer
- human papillomavirus (HPV)
- metabolism
- estrogens
1. Introduction
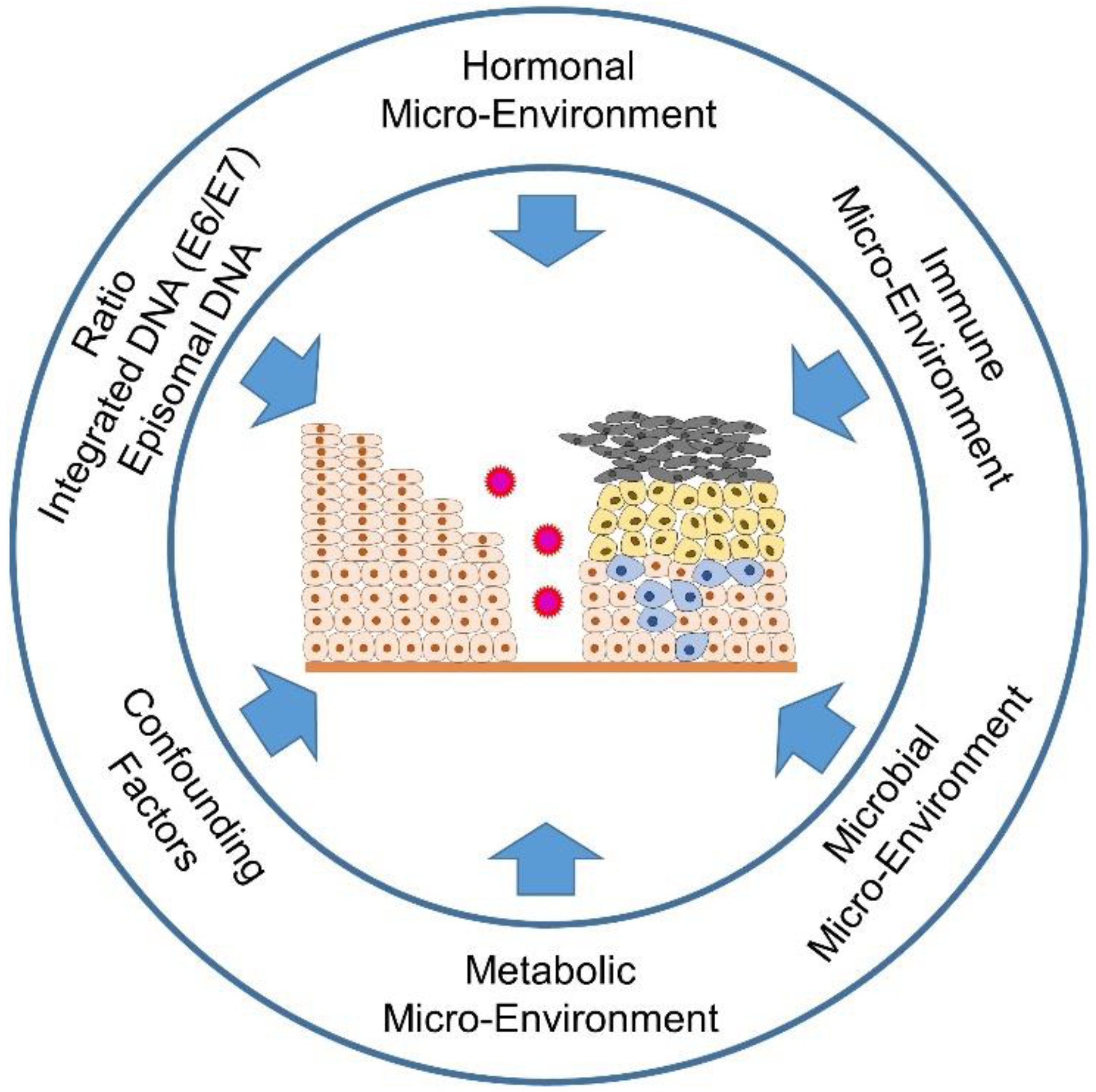
2. The Effects of the Hormone Microenvironment on Cervical Carcinogenesis
2.1. General Information about Sex Steroid Hormones, Their Biosynthesis and Their Localization in the Female Body
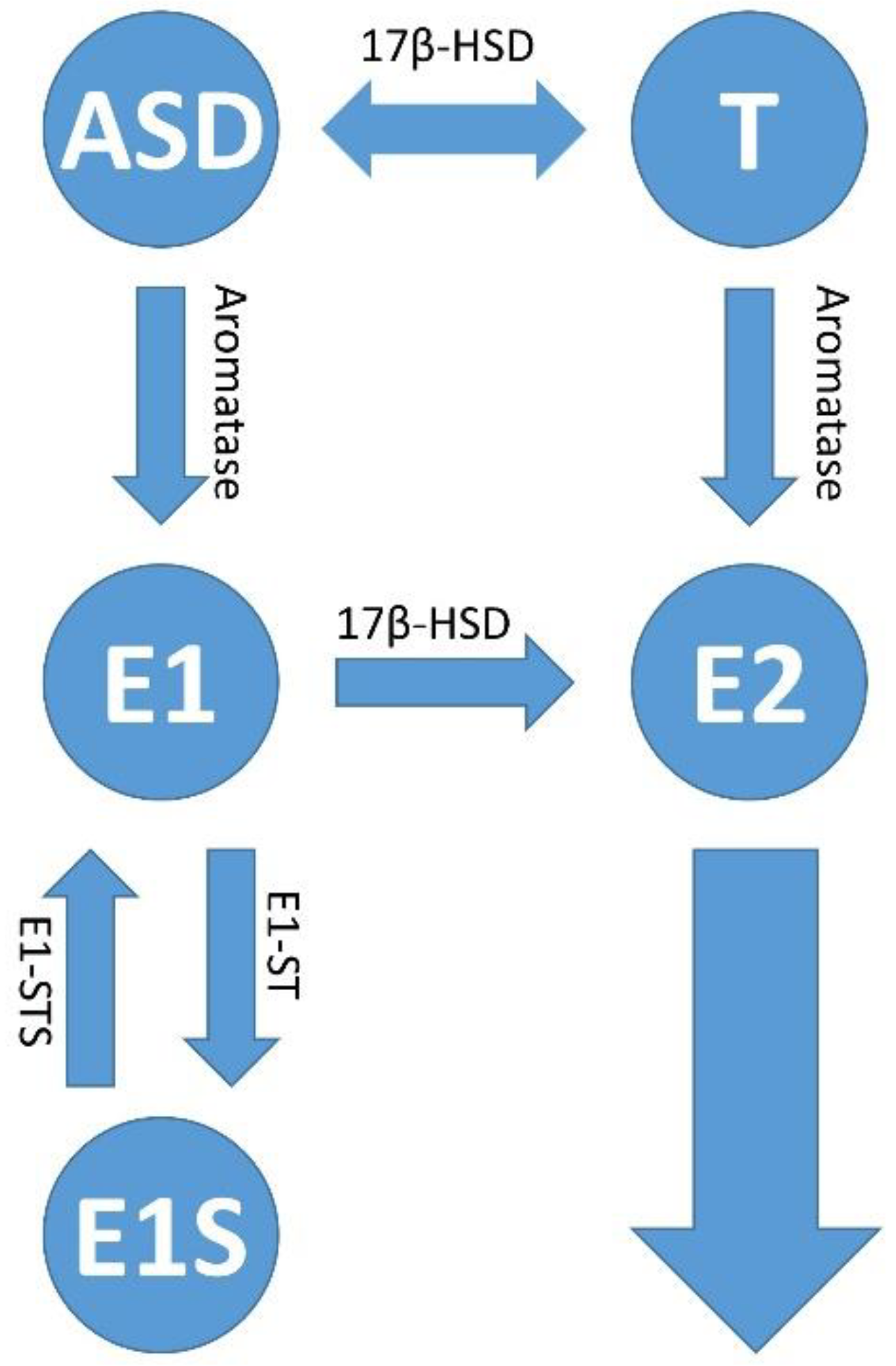
2.2. Estrogen Signaling
2.3. Influence of Estrogen Signaling on the Microenvironment of Cervical Intraepithelial Lesion, Cervical Pre-Cancer and in Tumor
2.3.1. Estrogen Distribution in the Tumor and the Surrounding Microenvironment
2.3.2. Different Estrogen Signaling Pathways Causing Both, Pro- and Anti-Tumorigenic Effects in HPV-Positive Lesions of the Cervical Epithelium
2.3.3. The Importance of the ERα for the Tumor Stroma and the Tumor Microenvironment in Relation to the Development of Precancerous Lesions up to the Invasive Form of Cervical Carcinoma
2.3.4. Estrogen Signaling in the Infiltrating Cells of the Immune Response in Cervical Carcinogenesis
2.4. The Effect of ER Antagonists in Modulating the Immune Microenvironment of the Premalignant Cervical Lesion and CxCa
This entry is adapted from the peer-reviewed paper 10.3390/ijms23095050
References
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; de Sanjosé, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers 2016, 2, 16086.
- De Vuyst, H.; Clifford, G.M.; Nascimento, M.C.; Madeleine, M.M.; Franceschi, S. Prevalence and type distribution of human papillomavirus in carcinoma and intraepithelial neoplasia of the vulva, vagina and anus: A meta-analysis. Int. J. Cancer 2009, 124, 1626–1636.
- Wittekindt, C.; Wagner, S.; Sharma, S.J.; Wurdemann, N.; Knuth, J.; Reder, H.; Klussmann, J.P. HPV—A different view on Head and Neck Cancer. Laryngorhinootologie 2018, 97, S48–S113.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Brianti, P.; De Flammineis, E.; Mercuri, S.R. Review of HPV-related diseases and cancers. New Microbiol. 2017, 40, 80–85.
- Chesson, H.W.; Dunne, E.F.; Hariri, S.; Markowitz, L.E. The estimated lifetime probability of acquiring human papillomavirus in the United States. Sex Transm. Dis. 2014, 41, 660–664.
- de Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 2017, 141, 664–670.
- Kang, S.D.; Chatterjee, S.; Alam, S.; Salzberg, A.C.; Milici, J.; van der Burg, S.H.; Meyers, C. Effect of Productive Human Papillomavirus 16 Infection on Global Gene Expression in Cervical Epithelium. J. Virol. 2018, 92, e01261-18.
- McBride, A.A.; Munger, K. Expert Views on HPV Infection. Viruses 2018, 10, 94.
- McLaughlin-Drubin, M.E.; Meyers, C. Evidence for the coexistence of two genital HPV types within the same host cell in vitro. Virology 2004, 321, 173–180.
- McLaughlin-Drubin, M.E.; Meyers, J.; Munger, K. Cancer associated human papillomaviruses. Curr. Opin. Virol. 2012, 2, 459–466.
- McLaughlin-Drubin, M.E.; Munger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208.
- James, C.D.; Morgan, I.M.; Bristol, M.L. The Relationship between Estrogen-Related Signaling and Human Papillomavirus Positive Cancers. Pathogens 2020, 9, 403.
- Marks, M.A.; Gravitt, P.E.; Burk, R.D.; Studentsov, Y.; Farzadegan, H.; Klein, S.L. Progesterone and 17beta-estradiol enhance regulatory responses to human papillomavirus type 16 virus-like particles in peripheral blood mononuclear cells from healthy women. Clin. Vaccine Immunol. 2010, 17, 609–617.
- Jayshree, R.S. The Immune Microenvironment in Human Papilloma Virus-Induced Cervical Lesions-Evidence for Estrogen as an Immunomodulator. Front. Cell. Infect. Microbiol. 2021, 11, 649815.
- Siegel, R.L.; Fedewa, S.A.; Miller, K.D.; Goding-Sauer, A.; Pinheiro, P.S.; Martinez-Tyson, D.; Jemal, A. Cancer statistics for Hispanics/Latinos, 2015. CA Cancer J. Clin. 2015, 65, 457–480.
- Viens, L.J.; Henley, S.J.; Watson, M.; Markowitz, L.E.; Thomas, C.C.; Thompson, T.D.; Razzaghi, H.; Saraiya, M. Human Papillomavirus-Associated Cancers—United States, 2008–2012. Morb. Mortal. Wkly. Rep. 2016, 65, 661–666.
- Hellberg, D. Sex steroids and cervical cancer. Anticancer Res. 2012, 32, 3045–3054.
- Li, B.; Zhang, L.; Zhao, J.; Tan, G.; Zhang, W.; Zhang, N.; Tian, J.; Qu, P. The value of cytokine levels in triage and risk prediction for women with persistent high-risk human papilloma virus infection of the cervix. Infect. Agents Cancer 2019, 14, 16.
- Nguyen, H.H.; Broker, T.R.; Chow, L.T.; Alvarez, R.D.; Vu, H.L.; Andrasi, J.; Brewer, L.R.; Jin, G.; Mestecky, J. Immune responses to human papillomavirus in genital tract of women with cervical cancer. Gynecol. Oncol. 2005, 96, 452–461.
- Samir, R.; Asplund, A.; Tot, T.; Pekar, G.; Hellberg, D. High-risk HPV infection and CIN grade correlates to the expression of c-myc, CD4+, FHIT, E-cadherin, Ki-67, and p16INK4a. J. Low. Genit. Tract Dis. 2011, 15, 280–286.
- Silins, I.; Kallings, I.; Dillner, J. Correlates of the spread of human papillomavirus infection. Cancer Epidemiol. Biomarkers Prev. 2000, 9, 953–959.
- Baker, J.M.; Al-Nakkash, L.; Herbst-Kralovetz, M.M. Estrogen-gut microbiome axis: Physiological and clinical implications. Maturitas 2017, 103, 45–53.
- Ratten, L.K.; Plummer, E.L.; Bradshaw, C.S.; Fairley, C.K.; Murray, G.L.; Garland, S.M.; Bateson, D.; Tachedjian, G.; Masson, L.; Vodstrcil, L.A. The Effect of Exogenous Sex Steroids on the Vaginal Microbiota: A Systematic Review. Front. Cell. Infect. Microbiol. 2021, 11, 732423.
- Zhou, Z.W.; Long, H.Z.; Cheng, Y.; Luo, H.Y.; Wen, D.D.; Gao, L.C. From Microbiome to Inflammation: The Key Drivers of Cervical Cancer. Front. Microbiol. 2021, 12, 767931.
- Wang, L.H.; Chen, L.R.; Chen, K.H. In Vitro and Vivo Identification, Metabolism and Action of Xenoestrogens: An Overview. Int. J. Mol. Sci. 2021, 22, 4013.
- Läsche, M.; Urban, H.; Gallwas, J.; Gründker, C. HPV and Other Microbiota; Who’s Good and Who’s Bad: Effects of the Microbial Environment on the Development of Cervical Cancer-A Non-Systematic Review. Cells 2021, 10, 714.
- Jia, M.; Dahlman-Wright, K.; Gustafsson, J. Estrogen receptor alpha and beta in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 557–568.
- Taneja, V. Sex Hormones Determine Immune Response. Front. Immunol. 2018, 9, 1931.
- Amador-Molina, A.; Hernández-Valencia, J.F.; Lamoyi, E.; Contreras-Paredes, A.; Lizano, M. Role of innate immunity against human papillomavirus (HPV) infections and effect of adjuvants in promoting specific immune response. Viruses 2013, 5, 2624–2642.
- Evans, M.R.; James, C.D.; Bristol, M.L.; Nulton, T.J.; Wang, X.; Kaur, N.; White, E.A.; Windle, B.; Morgan, I.M. Human Papillomavirus 16 E2 Regulates Keratinocyte Gene Expression Relevant to Cancer and the Viral Life Cycle. J. Virol. 2019, 93, e01941-18.
- Woodworth, C.D. HPV innate immunity. Front. Biosci. 2002, 7, d2058–d2071.
- Patel, S.; Homaei, A.; Raju, A.B.; Meher, B.R. Estrogen: The necessary evil for human health, and ways to tame it. Biomed. Pharmacother. 2018, 102, 403–411.
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 2017, 13, e1006211.
- Ferber, M.J.; Thorland, E.C.; Brink, A.A.; Rapp, A.K.; Phillips, L.A.; McGovern, R.; Gostout, B.S.; Cheung, T.H.; Chung, T.K.; Fu, W.Y.; et al. Preferential integration of human papillomavirus type 18 near the c-myc locus in cervical carcinoma. Oncogene 2003, 22, 7233–7242.
- Peter, M.; Rosty, C.; Couturier, J.; Radvanyi, F.; Teshima, H.; Sastre-Garau, X. MYC activation associated with the integration of HPV DNA at the MYC locus in genital tumors. Oncogene 2006, 25, 5985–5993.
- Smith, P.P.; Friedman, C.L.; Bryant, E.M.; McDougall, J.K. Viral integration and fragile sites in human papillomavirus-immortalized human keratinocyte cell lines. Genes Chromosomes Cancer 1992, 5, 150–157.
- Arias-Pulido, H.; Peyton, C.L.; Joste, N.E.; Vargas, H.; Wheeler, C.M. Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. J. Clin. Microbiol. 2006, 44, 1755–1762.
- Cannarella, R.; Condorelli, R.A.; Mongioì, L.M.; Barbagallo, F.; Calogero, A.E.; La Vignera, S. Effects of the selective estrogen receptor modulators for the treatment of male infertility: A systematic review and meta-analysis. Expert Opin. Pharmacother. 2019, 20, 1517–1525.
- Cauley, J.A. Estrogen and bone health in men and women. Steroids 2015, 99, 11–15.
- Dobbs, R.W.; Malhotra, N.R.; Greenwald, D.T.; Wang, A.Y.; Prins, G.S.; Abern, M.R. Estrogens and prostate cancer. Prostate Cancer Prostatic Dis. 2019, 22, 185–194.
- Harrison, R.F.; Bonnar, J. Clinical uses of estrogens. Pharmacol. Ther. 1980, 11, 451–467.
- Hasegawa, Y.; Itonaga, T.; Ikegawa, K.; Nishigaki, S.; Kawai, M.; Koga, E.; Sakakibara, H.; Ross, J.L. Ultra-low-dose estrogen therapy for female hypogonadism. Clin. Pediatr. Endocrinol. 2020, 29, 49–53.
- Knowlton, A.A.; Lee, A.R. Estrogen and the cardiovascular system. Pharmacol. Ther. 2012, 135, 54–70.
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell. Immunol. 2015, 294, 63–69.
- Lu, C.L.; Herndon, C. New roles for neuronal estrogen receptors. Neurogastroenterol. Motil. 2017, 29, e13121.
- Marquardt, R.M.; Kim, T.H.; Shin, J.H.; Jeong, J.W. Progesterone and Estrogen Signaling in the Endometrium: What Goes Wrong in Endometriosis? Int. J. Mol. Sci. 2019, 20, 3822.
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The role of estrogens in control of energy balance and glucose homeostasis. Endocr. Rev. 2013, 34, 309–338.
- Nakano, T.; Kadono, Y.; Iwamoto, H.; Yaegashi, H.; Iijima, M.; Kawaguchi, S.; Nohara, T.; Shigehara, K.; Izumi, K.; Mizokami, A. Therapeutic Effect of Ethinylestradiol in Castration-resistant Prostate Cancer. Anticancer Res. 2020, 40, 2291–2296.
- Pett, M.; Coleman, N. Integration of high-risk human papillomavirus: A key event in cervical carcinogenesis? J. Pathol. 2007, 212, 356–367.
- Regidor, P.A. Clinical relevance in present day hormonal contraception. Horm. Mol. Biol. Clin. Investig. 2018, 37.
- Simpson, E.R. Sources of estrogen and their importance. J. Steroid Biochem. Mol. Biol. 2003, 86, 225–230.
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170.
- Hariri, L.; Rehman, A. Estradiol. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022.
- Caroccia, B.; Seccia, T.M.; Barton, M.; Rossi, G.P. Estrogen Signaling in the Adrenal Cortex: Implications for Blood Pressure Sex Differences. Hypertension 2016, 68, 840–848.
- Nelson, L.R.; Bulun, S.E. Estrogen production and action. J. Am. Acad. Dermatol. 2001, 45, S116–S124.
- Wasada, T.; Akamine, Y.; Kato, K.; Ibayashi, H.; Nomura, Y. Adrenal contribution to circulating estrogens in woman. Endocrinol. Jpn 1978, 25, 123–128.
- Barakat, R.; Oakley, O.; Kim, H.; Jin, J.; Ko, C.J. Extra-gonadal sites of estrogen biosynthesis and function. BMB Rep. 2016, 49, 488–496.
- Cui, J.; Shen, Y.; Li, R. Estrogen synthesis and signaling pathways during aging: From periphery to brain. Trends Mol. Med. 2013, 19, 197–209.
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400.
- Marchesi, J.R.; Ravel, J. The vocabulary of microbiome research: A proposal. Microbiome 2015, 3, 31.
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533.
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359.
- Plottel, C.S.; Blaser, M.J. Microbiome and malignancy. Cell Host Microbe 2011, 10, 324–335.
- Gruber, C.J.; Tschugguel, W.; Schneeberger, C.; Huber, J.C. Production and actions of estrogens. N. Engl. J. Med. 2002, 346, 340–352.
- Zhu, B.T.; Han, G.Z.; Shim, J.Y.; Wen, Y.; Jiang, X.R. Quantitative structure-activity relationship of various endogenous estrogen metabolites for human estrogen receptor alpha and beta subtypes: Insights into the structural determinants favoring a differential subtype binding. Endocrinology 2006, 147, 4132–4150.
- Raftogianis, R.; Creveling, C.; Weinshilboum, R.; Weisz, J. Estrogen metabolism by conjugation. J. Natl. Cancer Inst. Monogr. 2000, 2000, 113–124.
- Dabek, M.; McCrae, S.I.; Stevens, V.J.; Duncan, S.H.; Louis, P. Distribution of beta-glucosidase and beta-glucuronidase activity and of beta-glucuronidase gene gus in human colonic bacteria. FEMS Microbiol. Ecol. 2008, 66, 487–495.
- Chen, K.L.; Madak-Erdogan, Z. Estrogen and Microbiota Crosstalk: Should We Pay Attention? Trends Endocrinol. Metab. 2016, 27, 752–755.
- Ervin, S.M.; Li, H.; Lim, L.; Roberts, L.R.; Liang, X.; Mani, S.; Redinbo, M.R. Gut microbial β-glucuronidases reactivate estrogens as components of the estrobolome that reactivate estrogens. J. Biol. Chem. 2019, 294, 18586–18599.
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front Cell Infect. Microbiol. 2018, 8, 13.
- Talib, W.H. Melatonin and Cancer Hallmarks. Molecules 2018, 23, 518.
- Blask, D.E.; Hill, S.M. Effects of melatonin on cancer: Studies on MCF-7 human breast cancer cells in culture. J. Neural Transm. Suppl. 1986, 21, 433–449.
- Hill, S.M.; Blask, D.E. Effects of the pineal hormone melatonin on the proliferation and morphological characteristics of human breast cancer cells (MCF-7) in culture. Cancer Res. 1988, 48, 6121–6126.
- Gonzalez, A.; Cos, S.; Martinez-Campa, C.; Alonso-Gonzalez, C.; Sanchez-Mateos, S.; Mediavilla, M.D.; Sanchez-Barcelo, E.J. Selective estrogen enzyme modulator actions of melatonin in human breast cancer cells. J. Pineal Res. 2008, 45, 86–92.
- Martínez-Campa, C.; González, A.; Mediavilla, M.D.; Alonso-González, C.; Alvarez-García, V.; Sánchez-Barceló, E.J.; Cos, S. Melatonin inhibits aromatase promoter expression by regulating cyclooxygenases expression and activity in breast cancer cells. Br. J. Cancer 2009, 101, 1613–1619.
- Somasundaram, A.; Rothenberger, N.J.; Stabile, L.P. The Impact of Estrogen in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1277, 33–52.
- Arbeit, J.M.; Howley, P.M.; Hanahan, D. Chronic estrogen-induced cervical and vaginal squamous carcinogenesis in human papillomavirus type 16 transgenic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 2930–2935.
- Brake, T.; Lambert, P.F. Estrogen contributes to the onset, persistence, and malignant progression of cervical cancer in a human papillomavirus-transgenic mouse model. Proc. Natl. Acad. Sci. USA 2005, 102, 2490–2495.
- Chung, S.H.; Lambert, P.F. Prevention and treatment of cervical cancer in mice using estrogen receptor antagonists. Proc. Natl. Acad. Sci. USA 2009, 106, 19467–19472.
- Muñoz, N.; Castellsagué, X.; Berrington de González, A.; Gissmann, L. Chapter 1: HPV in the etiology of human cancer. Vaccine 2006, 24 (Suppl. 3), S1–S10.
- Liarte, S.; Alonso-Romero, J.L.; Nicolás, F.J. SIRT1 and Estrogen Signaling Cooperation for Breast Cancer Onset and Progression. Front. Endocrinol. 2018, 9, 552.
- James, C.D.; Das, D.; Morgan, E.L.; Otoa, R.; Macdonald, A.; Morgan, I.M. Werner Syndrome Protein (WRN) Regulates Cell Proliferation and the Human Papillomavirus 16 Life Cycle during Epithelial Differentiation. mSphere 2020, 5, e00858-20.
- Jensen, K.E.; Schmiedel, S.; Norrild, B.; Frederiksen, K.; Iftner, T.; Kjaer, S.K. Parity as a cofactor for high-grade cervical disease among women with persistent human papillomavirus infection: A 13-year follow-up. Br. J. Cancer 2013, 108, 234–239.
- Moreno, V.; Bosch, F.X.; Muñoz, N.; Meijer, C.J.; Shah, K.V.; Walboomers, J.M.; Herrero, R.; Franceschi, S. Effect of oral contraceptives on risk of cervical cancer in women with human papillomavirus infection: The IARC multicentric case-control study. Lancet 2002, 359, 1085–1092.
- Muñoz, N.; Franceschi, S.; Bosetti, C.; Moreno, V.; Herrero, R.; Smith, J.S.; Shah, K.V.; Meijer, C.J.; Bosch, F.X. Role of parity and human papillomavirus in cervical cancer: The IARC multicentric case-control study. Lancet 2002, 359, 1093–1101.
- Rinaldi, S.; Plummer, M.; Biessy, C.; Castellsagué, X.; Overvad, K.; Krüger Kjær, S.; Tjønneland, A.; Clavel-Chapelon, F.; Chabbert-Buffet, N.; Mesrine, S.; et al. Endogenous sex steroids and risk of cervical carcinoma: Results from the EPIC study. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 2532–2540.
- Roura, E.; Travier, N.; Waterboer, T.; de Sanjosé, S.; Bosch, F.X.; Pawlita, M.; Pala, V.; Weiderpass, E.; Margall, N.; Dillner, J.; et al. The Influence of Hormonal Factors on the Risk of Developing Cervical Cancer and Pre-Cancer: Results from the EPIC Cohort. PLoS ONE 2016, 11, e0147029.
- Marks, M.; Klein, S.; Gravitt, P.; Feinstone, H. Hormonal contraception and HPV: A tale of differing and overlapping mechanisms. Open Access J. Contracept. 2011, 2, 161–174.
- Marks, M.A.; Viscidi, R.P.; Chang, K.; Silver, M.; Burke, A.; Howard, R.; Gravitt, P.E. Differences in the concentration and correlation of cervical immune markers among HPV positive and negative perimenopausal women. Cytokine 2011, 56, 798–803.
- Eibye, S.; Krüger Kjær, S.; Nielsen, T.S.; Mellemkjær, L. Mortality Among Women With Cervical Cancer During or Shortly After a Pregnancy in Denmark 1968 to 2006. Int. J. Gynecol. Cancer 2016, 26, 951–958.
- Lønning, P.E.; Haynes, B.P.; Straume, A.H.; Dunbier, A.; Helle, H.; Knappskog, S.; Dowsett, M. Exploring breast cancer estrogen disposition: The basis for endocrine manipulation. Clin. Cancer Res. 2011, 17, 4948–4958.
- Adurthi, S.; Kumar, M.M.; Vinodkumar, H.S.; Mukherjee, G.; Krishnamurthy, H.; Acharya, K.K.; Bafna, U.D.; Uma, D.K.; Abhishekh, B.; Krishna, S.; et al. Oestrogen Receptor-α binds the FOXP3 promoter and modulates regulatory T-cell function in human cervical cancer. Sci. Rep. 2017, 7, 17289.
- Kumar, M.M.; Davuluri, S.; Poojar, S.; Mukherjee, G.; Bajpai, A.K.; Bafna, U.D.; Devi, U.K.; Kallur, P.P.; Kshitish, A.K.; Jayshree, R.S. Role of estrogen receptor alpha in human cervical cancer-associated fibroblasts: A transcriptomic study. Tumor Biol. 2016, 37, 4409–4420.
- Nair, H.B.; Luthra, R.; Kirma, N.; Liu, Y.G.; Flowers, L.; Evans, D.; Tekmal, R.R. Induction of aromatase expression in cervical carcinomas: Effects of endogenous estrogen on cervical cancer cell proliferation. Cancer Res. 2005, 65, 11164–11173.
- Fournier, M.A.; Poirier, D. Estrogen formation in endometrial and cervix cancer cell lines: Involvement of aromatase, steroid sulfatase and 17beta-hydroxysteroid dehydrogenases (types 1, 5, 7 and 12). Mol. Cell. Endocrinol. 2009, 301, 142–145.
- Chen, Y.H.; Huang, L.H.; Chen, T.M. Differential effects of progestins and estrogens on long control regions of human papillomavirus types 16 and 18. Biochem. Biophys. Res. Commun. 1996, 224, 651–659.
- Kim, C.J.; Um, S.J.; Kim, T.Y.; Kim, E.J.; Park, T.C.; Kim, S.J.; Namkoong, S.E.; Park, J.S. Regulation of cell growth and HPV genes by exogenous estrogen in cervical cancer cells. Int. J. Gynecol. Cancer 2000, 10, 157–164.
- Mitrani-Rosenbaum, S.; Tsvieli, R.; Tur-Kaspa, R. Oestrogen stimulates differential transcription of human papillomavirus type 16 in SiHa cervical carcinoma cells. J. Gen. Virol. 1989, 70 Pt 8, 2227–2232.
- Ruutu, M.; Wahlroos, N.; Syrjänen, K.; Johansson, B.; Syrjänen, S. Effects of 17beta-estradiol and progesterone on transcription of human papillomavirus 16 E6/E7 oncogenes in CaSki and SiHa cell lines. Int. J. Gynecol. Cancer 2006, 16, 1261–1268.
- Bristol, M.L.; James, C.D.; Wang, X.; Fontan, C.T.; Morgan, I.M. Estrogen Attenuates the Growth of Human Papillomavirus-Positive Epithelial Cells. mSphere 2020, 5, e00049-20.
- Li, D.; Chen, J.; Ai, Y.; Gu, X.; Li, L.; Che, D.; Jiang, Z.; Chen, S.; Huang, H.; Wang, J.; et al. Estrogen-Related Hormones Induce Apoptosis by Stabilizing Schlafen-12 Protein Turnover. Mol. Cell 2019, 75, 1103–1116.e9.
- Lamb, H.M.; Hardwick, J.M. The Dark Side of Estrogen Stops Translation to Induce Apoptosis. Mol. Cell 2019, 75, 1087–1089.
- Park, J.S.; Rhyu, J.W.; Kim, C.J.; Kim, H.S.; Lee, S.Y.; Kwon, Y.I.; Namkoong, S.E.; Sin, H.S.; Um, S.J. Neoplastic change of squamo-columnar junction in uterine cervix and vaginal epithelium by exogenous estrogen in hpv-18 URR E6/E7 transgenic mice. Gynecol. Oncol. 2003, 89, 360–368.
- Chung, S.H.; Franceschi, S.; Lambert, P.F. Estrogen and ERalpha: Culprits in cervical cancer? Trends Endocrinol. Metab. 2010, 21, 504–511.
- Chung, S.H.; Wiedmeyer, K.; Shai, A.; Korach, K.S.; Lambert, P.F. Requirement for estrogen receptor alpha in a mouse model for human papillomavirus-associated cervical cancer. Cancer Res. 2008, 68, 9928–9934.
- Riley, R.R.; Duensing, S.; Brake, T.; Münger, K.; Lambert, P.F.; Arbeit, J.M. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003, 63, 4862–4871.
- Spurgeon, M.E.; den Boon, J.A.; Horswill, M.; Barthakur, S.; Forouzan, O.; Rader, J.S.; Beebe, D.J.; Roopra, A.; Ahlquist, P.; Lambert, P.F. Human papillomavirus oncogenes reprogram the cervical cancer microenvironment independently of and synergistically with estrogen. Proc. Natl. Acad. Sci. USA 2017, 114, E9076–E9085.
- Coelho, F.R.; Prado, J.C.; Pereira Sobrinho, J.S.; Hamada, G.; Landman, G.; Pinto, C.A.; Nonogaki, S.; Villa, L.L. Estrogen and progesterone receptors in human papilloma virus-related cervical neoplasia. Braz. J. Med. Biol. Res. 2004, 37, 83–88.
- den Boon, J.A.; Pyeon, D.; Wang, S.S.; Horswill, M.; Schiffman, M.; Sherman, M.; Zuna, R.E.; Wang, Z.; Hewitt, S.M.; Pearson, R.; et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E3255–E3264.
- Konishi, I.; Fujii, S.; Nonogaki, H.; Nanbu, Y.; Iwai, T.; Mori, T. Immunohistochemical analysis of estrogen receptors, progesterone receptors, Ki-67 antigen, and human papillomavirus DNA in normal and neoplastic epithelium of the uterine cervix. Cancer 1991, 68, 1340–1350.
- López-Romero, R.; Garrido-Guerrero, E.; Rangel-López, A.; Manuel-Apolinar, L.; Piña-Sánchez, P.; Lazos-Ochoa, M.; Mantilla-Morales, A.; Bandala, C.; Salcedo, M. The cervical malignant cells display a down regulation of ER-α but retain the ER-β expression. Int. J. Clin. Exp. Pathol. 2013, 6, 1594–1602.
- Nonogaki, H.; Fujii, S.; Konishi, I.; Nanbu, Y.; Ozaki, S.; Ishikawa, Y.; Mori, T. Estrogen receptor localization in normal and neoplastic epithelium of the uterine cervix. Cancer 1990, 66, 2620–2627.
- Zhai, Y.; Bommer, G.T.; Feng, Y.; Wiese, A.B.; Fearon, E.R.; Cho, K.R. Loss of estrogen receptor 1 enhances cervical cancer invasion. Am. J. Pathol. 2010, 177, 884–895.
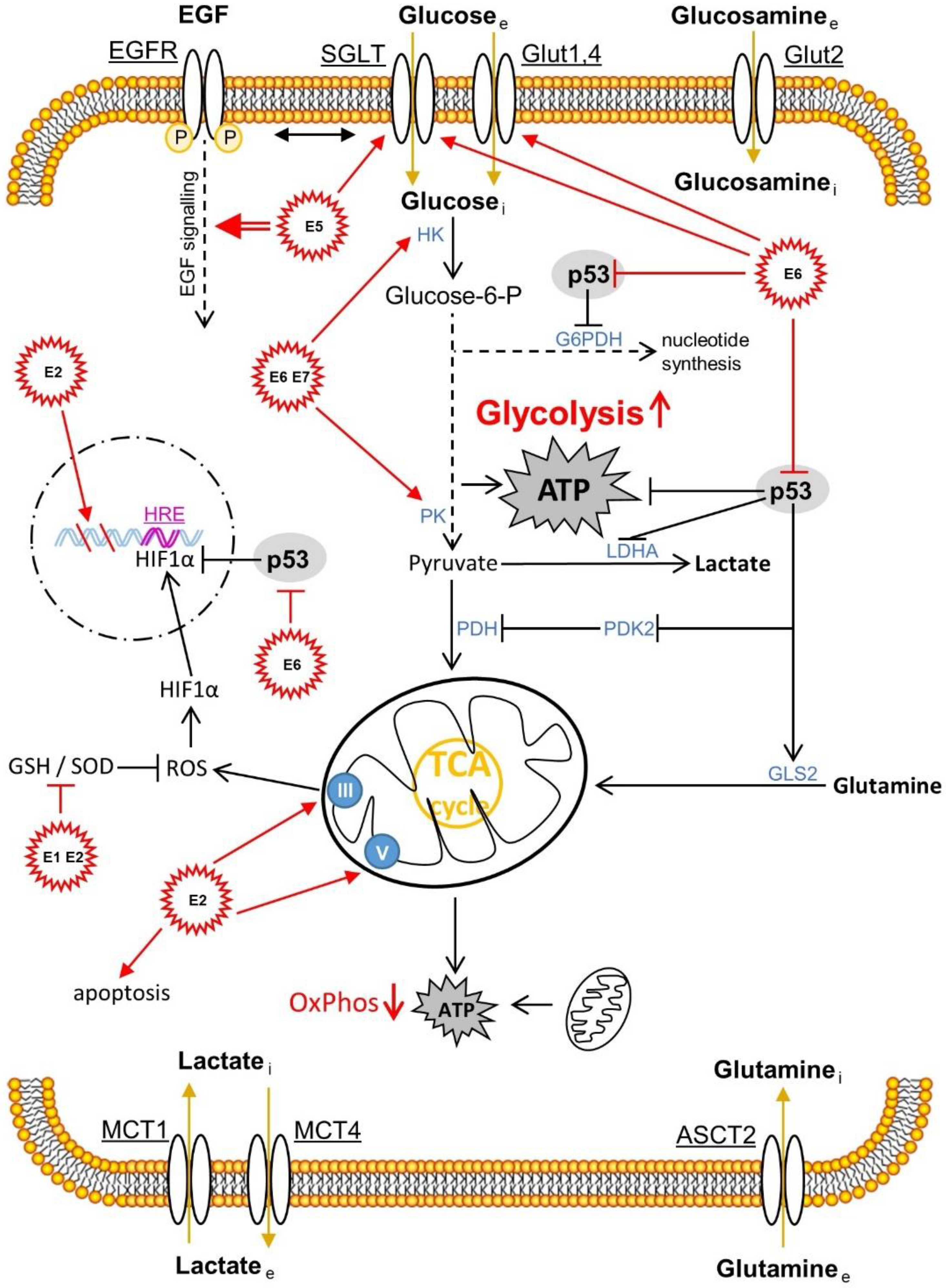
- Läsche, M.; Emons, G.; Gründker, C. Shedding New Light on Cancer Metabolism: A Metabolic Tightrope Between Life and Death. Front. Oncol. 2020, 10, 409.
- De Nola, R.; Menga, A.; Castegna, A.; Loizzi, V.; Ranieri, G.; Cicinelli, E.; Cormio, G. The Crowded Crosstalk between Cancer Cells and Stromal Microenvironment in Gynecological Malignancies: Biological Pathways and Therapeutic Implication. Int. J. Mol. Sci. 2019, 20, 2401.
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437.
- Kozasa, K.; Mabuchi, S.; Matsumoto, Y.; Kuroda, H.; Yokoi, E.; Komura, N.; Kawano, M.; Takahashi, R.; Sasano, T.; Shimura, K.; et al. Estrogen stimulates female cancer progression by inducing myeloid-derived suppressive cells: Investigations on pregnant and non-pregnant experimental models. Oncotarget 2019, 10, 1887–1902.
- Kwasniewska, A.; Postawski, K.; Gozdzicka-Jozefiak, A.; Kwasniewski, W.; Grywalska, E.; Zdunek, M.; Korobowicz, E. Estrogen and progesterone receptor expression in HPV-positive and HPV-negative cervical carcinomas. Oncol. Rep. 2011, 26, 153–160.
- Mosny, D.S.; Herholz, J.; Degen, W.; Bender, H.G. Immunohistochemical investigations of steroid receptors in normal and neoplastic squamous epithelium of the uterine cervix. Gynecol. Oncol. 1989, 35, 373–377.
- Chung, S.H.; Shin, M.K.; Korach, K.S.; Lambert, P.F. Requirement for stromal estrogen receptor alpha in cervical neoplasia. Horm. Cancer 2013, 4, 50–59.
- Spurgeon, M.E.; Lambert, P.F. Human Papillomavirus and the Stroma: Bidirectional Crosstalk during the Virus Life Cycle and Carcinogenesis. Viruses 2017, 9, 219.
- Son, J.; Park, Y.; Chung, S.H. Epithelial oestrogen receptor α is dispensable for the development of oestrogen-induced cervical neoplastic diseases. J. Pathol. 2018, 245, 147–152.
- Svoronos, N.; Perales-Puchalt, A.; Allegrezza, M.J.; Rutkowski, M.R.; Payne, K.K.; Tesone, A.J.; Nguyen, J.M.; Curiel, T.J.; Cadungog, M.G.; Singhal, S.; et al. Tumor Cell-Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov. 2017, 7, 72–85.
- Polanczyk, M.J.; Carson, B.D.; Subramanian, S.; Afentoulis, M.; Vandenbark, A.A.; Ziegler, S.F.; Offner, H. Cutting edge: Estrogen drives expansion of the CD4+CD25+ regulatory T cell compartment. J. Immunol. 2004, 173, 2227–2230.
- Yang, X.F. Factors regulating apoptosis and homeostasis of CD4+ CD25(high) FOXP3+ regulatory T cells are new therapeutic targets. Front. Biosci. 2008, 13, 1472–1499.
- Cao, X.; Cai, S.F.; Fehniger, T.A.; Song, J.; Collins, L.I.; Piwnica-Worms, D.R.; Ley, T.J. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 2007, 27, 635–646.
- Polanczyk, M.J.; Hopke, C.; Vandenbark, A.A.; Offner, H. Estrogen-mediated immunomodulation involves reduced activation of effector T cells, potentiation of Treg cells, and enhanced expression of the PD-1 costimulatory pathway. J. Neurosci. Res. 2006, 84, 370–378.
- Polanczyk, M.J.; Hopke, C.; Vandenbark, A.A.; Offner, H. Treg suppressive activity involves estrogen-dependent expression of programmed death-1 (PD-1). Int. Immunol. 2007, 19, 337–343.
- Prieto, G.A.; Rosenstein, Y. Oestradiol potentiates the suppressive function of human CD4 CD25 regulatory T cells by promoting their proliferation. Immunology 2006, 118, 58–65.
- Valor, L.; Teijeiro, R.; Aristimuño, C.; Faure, F.; Alonso, B.; de Andrés, C.; Tejera, M.; López-Lazareno, N.; Fernández-Cruz, E.; Sánchez-Ramón, S. Estradiol-dependent perforin expression by human regulatory T-cells. Eur. J. Clin. Investig. 2011, 41, 357–364.
- Yates, M.A.; Li, Y.; Chlebeck, P.J.; Offner, H. GPR30, but not estrogen receptor-alpha, is crucial in the treatment of experimental autoimmune encephalomyelitis by oral ethinyl estradiol. BMC Immunol. 2010, 11, 20.
- Traboulsi, T.; El Ezzy, M.; Gleason, J.L.; Mader, S. Antiestrogens: Structure-activity relationships and use in breast cancer treatment. J. Mol. Endocrinol. 2017, 58, R15–R31.
- Khan, M.S.; Singh, P.; Azhar, A.; Naseem, A.; Rashid, Q.; Kabir, M.A.; Jairajpuri, M.A. Serpin Inhibition Mechanism: A Delicate Balance between Native Metastable State and Polymerization. J. Amino Acids 2011, 2011, 606797.
- Medema, J.P.; de Jong, J.; Peltenburg, L.T.; Verdegaal, E.M.; Gorter, A.; Bres, S.A.; Franken, K.L.; Hahne, M.; Albar, J.P.; Melief, C.J.; et al. Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 11515–11520.
- Munguía-Moreno, J.A.; Díaz-Chavéz, J.; García-Villa, E.; Albino-Sanchez, M.E.; Mendoza-Villanueva, D.; Ocadiz-Delgado, R.; Bonilla-Delgado, J.; Marín-Flores, A.; Cortés-Malagón, E.M.; Alvarez-Rios, E.; et al. Early synergistic interactions between the HPV16-E7 oncoprotein and 17β-oestradiol for repressing the expression of Granzyme B in a cervical cancer model. Int. J. Oncol. 2018, 53, 579–591.
- Hernandez-Pigeon, H.; Jean, C.; Charruyer, A.; Haure, M.J.; Baudouin, C.; Charveron, M.; Quillet-Mary, A.; Laurent, G. UVA induces granzyme B in human keratinocytes through MIF: Implication in extracellular matrix remodeling. J. Biol. Chem. 2007, 282, 8157–8164.
- Parkinson, L.G.; Toro, A.; Zhao, H.; Brown, K.; Tebbutt, S.J.; Granville, D.J. Granzyme B mediates both direct and indirect cleavage of extracellular matrix in skin after chronic low-dose ultraviolet light irradiation. Aging Cell 2015, 14, 67–77.
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910.
- Jiang, X.; Orr, B.A.; Kranz, D.M.; Shapiro, D.J. Estrogen induction of the granzyme B inhibitor, proteinase inhibitor 9, protects cells against apoptosis mediated by cytotoxic T lymphocytes and natural killer cells. Endocrinology 2006, 147, 1419–1426.
- Svensson, S.; Abrahamsson, A.; Rodriguez, G.V.; Olsson, A.K.; Jensen, L.; Cao, Y.; Dabrosin, C. CCL2 and CCL5 Are Novel Therapeutic Targets for Estrogen-Dependent Breast Cancer. Clin. Cancer Res. 2015, 21, 3794–3805.
- deGraffenried, L.A.; Hilsenbeck, S.G.; Fuqua, S.A. Sp1 is essential for estrogen receptor alpha gene transcription. J. Steroid Biochem. Mol. Biol. 2002, 82, 7–18.
- Marino, M.; Galluzzo, P.; Ascenzi, P. Estrogen signaling multiple pathways to impact gene transcription. Curr. Genom. 2006, 7, 497–508.
- Webb, P.; Nguyen, P.; Valentine, C.; Lopez, G.N.; Kwok, G.R.; McInerney, E.; Katzenellenbogen, B.S.; Enmark, E.; Gustafsson, J.A.; Nilsson, S.; et al. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol. Endocrinol. 1999, 13, 1672–1685.
- Hsieh, C.J.; Hong, M.K.; Chen, P.C.; Wang, J.H.; Chu, T.Y. Antiestrogen use reduces risk of cervical neoplasia in breast cancer patients: A population-based study. Oncotarget 2017, 8, 29361–29369.
- Generali, D.; Bates, G.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Bersiga, A.; Allevi, G.; Milani, M.; Aguggini, S.; et al. Immunomodulation of FOXP3+ regulatory T cells by the aromatase inhibitor letrozole in breast cancer patients. Clin. Cancer Res. 2009, 15, 1046–1051.
- Ray, A.; Ficek, M. Immunomodulatory effects of anti-estrogenic drugs. Acta Pharm. 2012, 62, 141–155.
- Polese, B.; Gridelet, V.; Araklioti, E.; Martens, H.; Perrier d’Hauterive, S.; Geenen, V. The Endocrine Milieu and CD4 T-Lymphocyte Polarization during Pregnancy. Front. Endocrinol. 2014, 5, 106.
- Purohit, A.; Foster, P.A. Steroid sulfatase inhibitors for estrogen- and androgen-dependent cancers. J. Endocrinol. 2012, 212, 99–110.
- Secky, L.; Svoboda, M.; Klameth, L.; Bajna, E.; Hamilton, G.; Zeillinger, R.; Jäger, W.; Thalhammer, T. The sulfatase pathway for estrogen formation: Targets for the treatment and diagnosis of hormone-associated tumors. J. Drug Deliv. 2013, 2013, 957605.