 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Emre Bektik | -- | 2538 | 2022-05-05 20:38:29 | | | |
| 2 | Jessie Wu | -6 word(s) | 2532 | 2022-05-06 04:17:13 | | |
Video Upload Options
Coronary artery disease is the most common form of cardiovascular diseases, resulting in the loss of cardiomyocytes (CM) at the site of ischemic injury. To compensate for the loss of CMs, cardiac fibroblasts quickly respond to injury and initiate cardiac remodeling in an injured heart. In the remodeling process, cardiac fibroblasts proliferate and differentiate into myofibroblasts, which secrete extracellular matrix to support the intact structure of the heart, and eventually differentiate into matrifibrocytes to form chronic scar tissue. Discovery of direct cardiac reprogramming offers a promising therapeutic strategy to prevent/attenuate this pathologic remodeling and replace the cardiac fibrotic scar with myocardium in situ. Since the first discovery in 2010, many progresses have been made to improve the efficiency and efficacy of reprogramming by understanding the mechanisms and signaling pathways that are activated during direct cardiac reprogramming.
1. Direct Reprogramming of Mouse Fibroblasts into iCMs In Vitro
2. In Situ Reprogramming of iCMs in The Heart
3. Direct Cardiac Reprogramming of Human Fibroblasts
4. Mechanistical Understanding of Direct Cardiac Reprogramming
4.1. Activation of Signaling Pathways During Reprogramming
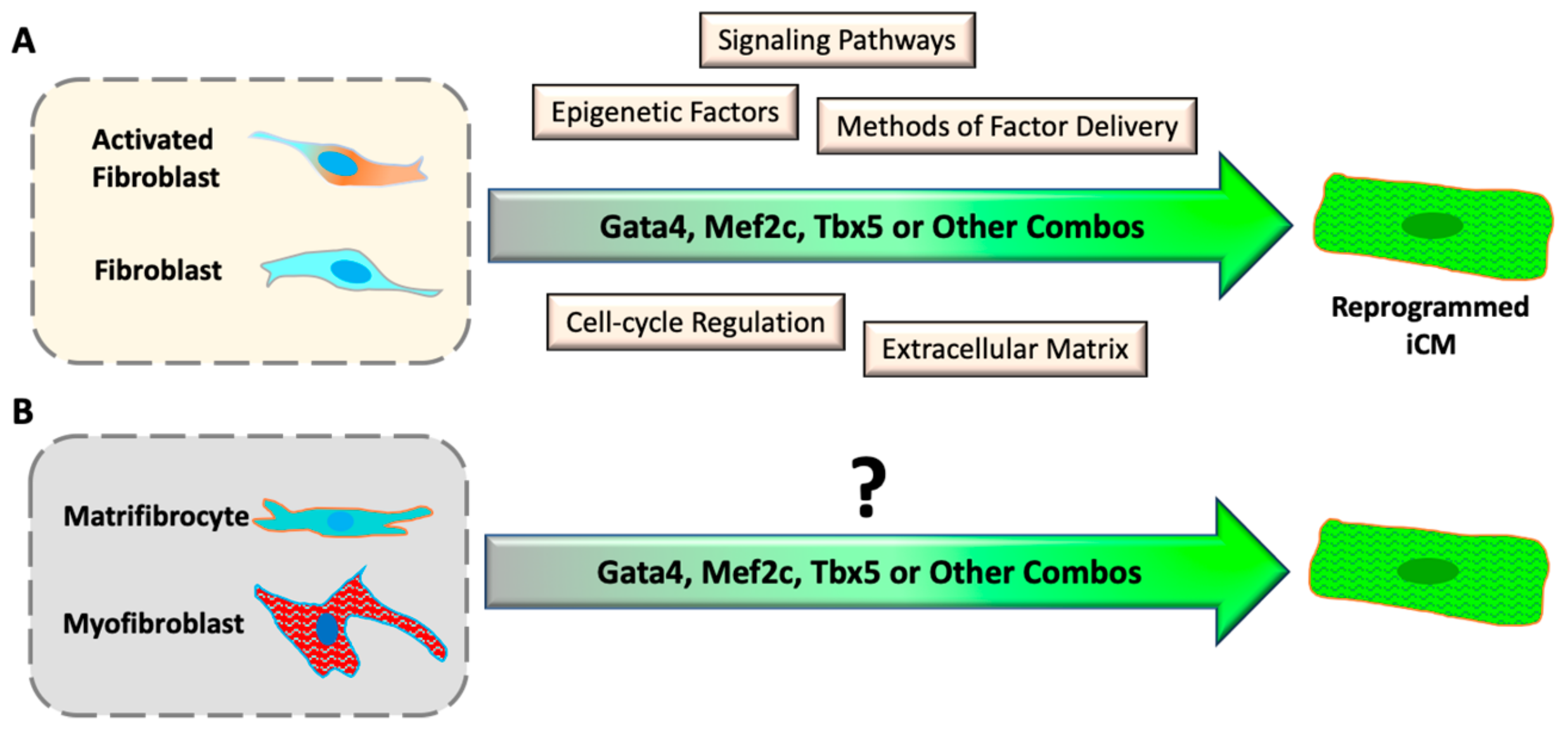
4.2. Epigenetic Barriers of Reprogramming
4.3. Cell-Cycle Regulation During Direct Cardiac Reprogramming
4.4. Modification of Extracellular Matrix
References
- Choi, J.; Costa, M.L.; Mermelstein, C.S.; Chagas, C.; Holtzer, S.; Holtzer, H. MyoD converts primary dermal fibroblasts, chondroblasts, smooth muscle, and retinal pigmented epithelial cells into striated mononucleated myoblasts and multinucleated myotubes. Proc. Natl. Acad. Sci. USA 1990, 87, 7988–7992.
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676.
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872.
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct Reprogramming of Fibroblasts into Functional Cardiomyocytes by Defined Factors. Cell 2010, 142, 375–386.
- Song, K.; Nam, Y.J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G.; et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604.
- Addis, R.C.; Ifkovits, J.L.; Pinto, F.; Kellam, L.D.; Esteso, P.; Rentschler, S.; Christoforou, N.; Epstein, J.A.; Gearhart, J.D. Optimization of direct fibroblast reprogramming to cardiomyocytes using calcium activity as a functional measure of success. J. Mol. Cell. Cardiol. 2013, 60, 97–106.
- Protze, S.; Khattak, S.; Poulet, C.; Lindemann, D.; Tanaka, E.M.; Ravens, U. A new approach to transcription factor screening for reprogramming of fibroblasts to cardiomyocyte-like cells. J. Mol. Cell. Cardiol. 2012, 53, 323–332.
- Hirai, H.; Katoku-Kikyo, N.; Keirstead, S.A.; Kikyo, N. Accelerated direct reprogramming of fibroblasts into cardiomyocyte-like cells with the MyoD transactivation domain. Cardiovasc. Res. 2013, 100, 105–113.
- Jayawardena, T.M.; Egemnazarov, B.; Finch, E.A.; Zhang, L.; Payne, J.A.; Pandya, K.; Zhang, Z.; Rosenberg, P.; Mirotsou, M.; Dzau, V.J. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ. Res. 2012, 110, 1465–1473.
- Muraoka, N.; Yamakawa, H.; Miyamoto, K.; Sadahiro, T.; Umei, T.; Isomi, M.; Nakashima, H.; Akiyama, M.; Wada, R.; Inagawa, K.; et al. MiR-133 promotes cardiac reprogramming by directly repressing Snai1 and silencing fibroblast signatures. EMBO J. 2014, 33, 1565–1581.
- Stone, N.R.; Gifford, C.A.; Thomas, R.; Pratt, K.J.B.; Samse-Knapp, K.; Mohamed, T.M.A.; Radzinsky, E.M.; Schricker, A.; Yu, P.; Ivey, K.N.; et al. Unique Transcription Factor Functions Regulate Epigenetic and Transcriptional Dynamics During Cardiac Reprogramming. bioRxiv 2019.
- Zhang, Z.; Zhang, A.D.; Kim, L.J.; Nam, Y.J. Ensuring expression of four core cardiogenic transcription factors enhances cardiac reprogramming. Sci. Rep. 2019, 9, 6362.
- Qian, L.; Huang, Y.; Spencer, C.I.; Foley, A.; Vedantham, V.; Liu, L.; Conway, S.J.; Fu, J.-d.D.; Srivastava, D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 2012, 485, 593–598.
- Srivastava, D.; Ieda, M.; Fu, J.; Qian, L. Cardiac repair with thymosin β4 and cardiac reprogramming factors. Ann. N. Y. Acad. Sci. 2012, 1270, 66–72.
- Jayawardena, T.M.; Finch, E.A.; Zhang, L.; Zhang, H.; Hodgkinson, C.P.; Pratt, R.E.; Rosenberg, P.B.; Mirotsou, M.; Dzau, V.J. MicroRNA induced cardiac reprogramming in vivo: Evidence for mature cardiac myocytes and improved cardiac function. Circ. Res. 2015, 116, 418–424.
- Inagawa, K.; Miyamoto, K.; Yamakawa, H.; Muraoka, N.; Sadahiro, T.; Umei, T.; Wada, R.; Katsumata, Y.; Kaneda, R.; Nakade, K.; et al. Induction of cardiomyocyte-like cells in infarct hearts by gene transfer of Gata4, Mef2c, and Tbx5. Circ. Res. 2012, 111, 1147–1156.
- Wang, L.; Liu, Z.; Yin, C.; Asfour, H.; Chen, O.; Li, Y.; Bursac, N.; Liu, J.; Qian, L. Stoichiometry of Gata4, Mef2c, and Tbx5 Influences the Efficiency and Quality of Induced Cardiac Myocyte Reprogramming. Circ. Res. 2015, 116, 237–244.
- Ma, H.; Wang, L.; Yin, C.; Liu, J.; Qian, L. In vivo cardiac reprogramming using an optimal single polycistronic construct. Cardiovasc. Res. 2015, 108, 217–219.
- Liu, Z.; Wang, L.; Welch, J.D.; Ma, H.; Zhou, Y.; Vaseghi, H.R.; Yu, S.; Wall, J.B.; Alimohamadi, S.; Zheng, M.; et al. Single-cell transcriptomics reconstructs fate conversion from fibroblast to cardiomyocyte. Nature 2017.
- Wada, R.; Muraoka, N.; Inagawa, K.; Yamakawa, H.; Miyamoto, K.; Sadahiro, T.; Umei, T.; Kaneda, R.; Suzuki, T.; Kamiya, K.; et al. Induction of human cardiomyocyte-like cells from fibroblasts by defined factors. Proc. Natl. Acad. Sci. USA 2013, 110, 12667–12672.
- Fu, J.-D.D.; Stone, N.R.; Liu, L.; Spencer, C.I.; Qian, L.; Hayashi, Y.; Delgado-Olguin, P.; Ding, S.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of human fibroblasts toward a cardiomyocyte-like state. Stem Cell Rep. 2013, 1, 235–247.
- Bektik, E.; Dennis, A.; Prasanna, P.; Madabhushi, A.; Fu, J.-D.D. Single cell qPCR reveals that additional HAND2 and microRNA-1 facilitate the early reprogramming progress of seven-factor-induced human myocytes. PLoS ONE 2017, 12, e0183000.
- Mohamed, T.M.A.; Stone, N.R.; Berry, E.C.; Radzinsky, E.; Huang, Y.; Pratt, K.; Ang, Y.-S.; Yu, P.; Wang, H.; Tang, S.; et al. Chemical Enhancement of In Vitro and In Vivo Direct Cardiac Reprogramming Clinical Perspective. Circulation 2016, 135, 978–995.
- Nam, Y.J.; Song, K.; Luo, X.; Daniel, E.; Lambeth, K.; West, K.; Hill, J.A.; DiMaio, J.M.; Baker, L.A.; Bassel-Duby, R.; et al. Reprogramming of human fibroblasts toward a cardiac fate. Proc. Natl. Acad. Sci. USA 2013, 110, 5588–5593.
- Ifkovits, J.L.; Addis, R.C.; Epstein, J.A.; Gearhart, J.D. Inhibition of TGFbeta signaling increases direct conversion of fibroblasts to induced cardiomyocytes. PLoS ONE 2014, 9, e89678.
- Zhao, Y.; Londono, P.; Cao, Y.; Sharpe, E.J.; Proenza, C.; O’Rourke, R.; Jones, K.L.; Jeong, M.Y.; Walker, L.A.; Buttrick, P.M.; et al. High-efficiency reprogramming of fibroblasts into cardiomyocytes requires suppression of pro-fibrotic signalling. Nat. Commun. 2015, 6, 8243.
- Bektik, E.; Dennis, A.; Pawlowski, G.; Zhou, C.; Maleski, D.; Takahashi, S.; Laurita, K.R.; Deschênes, I.; Fu, J.-D.D. S-phase Synchronization Facilitates the Early Progression of Induced-Cardiomyocyte Reprogramming through Enhanced Cell-Cycle Exit. Int. J. Mol. Sci. 2018, 19, 1364.
- Abad, M.; Hashimoto, H.; Zhou, H.; Morales, M.G.; Chen, B.; Bassel-Duby, R.; Olson, E.N. Notch Inhibition Enhances Cardiac Reprogramming by Increasing MEF2C Transcriptional Activity. Stem Cell Rep. 2017, 8, 548–560.
- Zhou, H.; Dickson, M.E.; Kim, M.S.; Bassel-Duby, R.; Olson, E.N. Akt1/protein kinase B enhances transcriptional reprogramming of fibroblasts to functional cardiomyocytes. Proc. Natl. Acad. Sci. USA 2015, 112, 11864–11869.
- Yamakawa, H.; Muraoka, N.; Miyamoto, K.; Sadahiro, T.; Isomi, M.; Haginiwa, S.; Kojima, H.; Umei, T.; Akiyama, M.; Kuishi, Y.; et al. Fibroblast Growth Factors and Vascular Endothelial Growth Factor Promote Cardiac Reprogramming under Defined Conditions. Stem Cell Rep. 2015, 5, 1128–1142.
- Zhou, H.; Morales, M.G.; Hashimoto, H.; Dickson, M.E.; Song, K.; Ye, W.; Kim, M.S.; Niederstrasser, H.; Wang, Z.; Chen, B.; et al. ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression. Genes Dev. 2017, 31, 1770–1783.
- Yijing, G.; Ienglam, L.; Shuo, T.; Wenbin, G.; Karatas, H.; Yangbing, L.; Shaomeng, W.; Liu, L.; Zhong, W. Enhancing Cardiac Reprogramming by Suppressing Specific C-C Chemokine Signaling Pathways. bioRxiv 2019.
- Muraoka, N.; Nara, K.; Tamura, F.; Kojima, H.; Yamakawa, H.; Sadahiro, T.; Miyamoto, K.; Isomi, M.; Haginiwa, S.; Tani, H.; et al. Role of cyclooxygenase-2-mediated prostaglandin E2-prostaglandin E receptor 4 signaling in cardiac reprogramming. Nat. Commun. 2019, 10, 674.
- Soufi, A.; Donahue, G.; Zaret, K.S. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 2012, 151, 994–1004.
- Chronis, C.; Fiziev, P.; Papp, B.; Butz, S.; Bonora, G.; Sabri, S.; Ernst, J.; Plath, K. Cooperative Binding of Transcription Factors Orchestrates Reprogramming. Cell 2017, 168, 442–459.
- Wapinski, O.L.; Vierbuchen, T.; Qu, K.; Lee, Q.Y.; Chanda, S.; Fuentes, D.R.; Giresi, P.G.; Ng, Y.H.; Marro, S.; Neff, N.F.; et al. Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell 2013, 155, 621–635.
- Chanda, S.; Ang, C.E.; Davila, J.; Pak, C.; Mall, M.; Lee, Q.Y.; Ahlenius, H.; Jung, S.W.; Sudhof, T.C.; Wernig, M. Generation of induced neuronal cells by the single reprogramming factor ASCL1. Stem Cell Rep. 2014, 3, 282–296.
- Liu, Z.; Chen, O.; Zheng, M.; Wang, L.; Zhou, Y.; Yin, C.; Liu, J.; Qian, L. Re-patterning of H3K27me3, H3K4me3 and DNA methylation during fibroblast conversion into induced cardiomyocytes. Stem Cell Res. 2016, 16, 507–518.
- Hirai, H.; Kikyo, N. Inhibitors of suppressive histone modification promote direct reprogramming of fibroblasts to cardiomyocyte-like cells. Cardiovasc. Res. 2014, 102, 188–190.
- Dal-Pra, S.; Hodgkinson, C.P.; Mirotsou, M.; Kirste, I.; Dzau, V.J. Demethylation of H3K27 Is Essential for the Induction of Direct Cardiac Reprogramming by miR Combo. Circ. Res. 2017, 120, 1403–1413.
- Zhou, Y.; Wang, L.; Vaseghi, H.R.; Liu, Z.; Lu, R.; Alimohamadi, S.; Yin, C.; Fu, J.-D.D.; Wang, G.G.; Liu, J.; et al. Bmi1 Is a Key Epigenetic Barrier to Direct Cardiac Reprogramming. Cell Stem Cell 2016, 18, 382–395.
- Hashimoto, H.; Wang, Z.; Garry, G.A.; Malladi, V.S.; Botten, G.A.; Ye, W.; Zhou, H.; Osterwalder, M.; Dickel, D.E.; Visel, A.; et al. Cardiac Reprogramming Factors Synergistically Activate Genome-wide Cardiogenic Stage-Specific Enhancers. Cell Stem Cell 2019.
- Donaghey, J.; Thakurela, S.; Charlton, J.; Chen, J.S.; Smith, Z.D.; Gu, H.; Pop, R.; Clement, K.; Stamenova, E.K.; Karnik, R.; et al. Genetic determinants and epigenetic effects of pioneer-factor occupancy. Nat. Genet. 2018, 50, 250–258.
- Souders, C.A.; Bowers, S.L.; Baudino, T.A. Cardiac fibroblast: The renaissance cell. Circ. Res. 2009, 105, 1164–1176.
- Brown, S.E.; Fraga, M.F.; Weaver, I.C.; Berdasco, M.; Szyf, M. Variations in DNA methylation patterns during the cell cycle of HeLa cells. Epigenetics 2007, 2, 54–65.
- Bou Kheir, T.; Lund, A.H. Epigenetic dynamics across the cell cycle. Essays Biochem. 2010, 48, 107–120.
- Zhou, Y.; Wang, L.; Liu, Z.; Alimohamadi, S.; Yin, C.; Liu, J.; Qian, L. Comparative Gene Expression Analyses Reveal Distinct Molecular Signatures between Differentially Reprogrammed Cardiomyocytes. Cell Rep. 2017, 20, 3014–3024.
- Zhou, Y.; Liu, Z.; Welch, J.D.; Gao, X.; Wang, L.; Garbutt, T.; Keepers, B.; Ma, H.; Prins, J.F.; Shen, W.; et al. Single-Cell Transcriptomic Analyses of Cell Fate Transitions during Human Cardiac Reprogramming. Cell Stem Cell 2019.
- Takeuchi, T. Regulation of cardiomyocyte proliferation during development and regeneration. Dev. Growth Differ. 2014, 56, 402–409.
- Chopra, A.; Lin, V.; McCollough, A.; Atzet, S.; Prestwich, G.D.; Wechsler, A.S.; Murray, M.E.; Oake, S.A.; Kresh, J.Y.; Janmey, P.A. Reprogramming cardiomyocyte mechanosensing by crosstalk between integrins and hyaluronic acid receptors. J. Biomech. 2012, 45, 824–831.
- Engler, A.J.; Carag-Krieger, C.; Johnson, C.P.; Raab, M.; Tang, H.Y.; Speicher, D.W.; Sanger, J.W.; Sanger, J.M.; Discher, D.E. Embryonic cardiomyocytes beat best on a matrix with heart-like elasticity: Scar-like rigidity inhibits beating. J. Cell Sci. 2008, 121, 3794–3802.
- Ribeiro, A.J.; Ang, Y.S.; Fu, J.D.; Rivas, R.N.; Mohamed, T.M.; Higgs, G.C.; Srivastava, D.; Pruitt, B.L. Contractility of single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. Proc. Natl. Acad. Sci. USA 2015, 112, 12705–12710.
- Sia, J.; Yu, P.; Srivastava, D.; Li, S. Effect of biophysical cues on reprogramming to cardiomyocytes. Biomaterials 2016, 103, 1–11.
- Li, Y.Z.; Dal-Pra, S.; Mirotsou, M.; Jayawardena, T.M.; Hodgkinson, C.P.; Bursac, N.; Dzau, V.J. Tissue-engineered 3-dimensional (3D) microenvironment enhances the direct reprogramming of fibroblasts into cardiomyocytes by microRNAs. Sci. Rep. 2016, 6.
- Zhang, S.; Sun, A.; Ma, H.; Yao, K.; Zhou, N.; Shen, L.; Zhang, C.; Zou, Y.; Ge, J. Infarcted myocardium-like stiffness contributes to endothelial progenitor lineage commitment of bone marrow mononuclear cells. J. Cell. Mol. Med. 2011, 15, 2245–2261.
- Voorhees, A.P.; DeLeon-Pennell, K.Y.; Ma, Y.; Halade, G.V.; Yabluchanskiy, A.; Iyer, R.P.; Flynn, E.; Cates, C.A.; Lindsey, M.L.; Han, H.C. Building a better infarct: Modulation of collagen cross-linking to increase infarct stiffness and reduce left ventricular dilation post-myocardial infarction. J. Mol. Cell. Cardiol. 2015, 85, 229–239.

