 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Emiliano Panieri | -- | 3437 | 2022-05-05 11:10:13 | | | |
| 2 | Vivi Li | + 88 word(s) | 3525 | 2022-05-05 11:56:59 | | |
Video Upload Options
Ischemic heart disease is a leading cause of death worldwide. Primarily, ischemia causes decreased oxygen supply, resulting in damage of the cardiac tissue. Naturally, reoxygenation has been recognized as the treatment of choice to recover blood flow through primary percutaneous coronary intervention. This treatment is the gold standard therapy to restore blood flow, but paradoxically it can also induce tissue injury. A number of different studies in animal models of acute myocardial infarction (AMI) suggest that ischemia-reperfusion injury (IRI) accounts for up to 50% of the final myocardial infarct size. Oxidative stress plays a critical role in the pathological process. Iron is an essential mineral required for a variety of vital biological functions but also has potentially toxic effects. A detrimental process induced by free iron is ferroptosis, a non-apoptotic type of programmed cell death. Accordingly, efforts to prevent ferroptosis in pathological settings have focused on the use of radical trapping antioxidants (RTAs), such as liproxstatin-1 (Lip-1). Hence, it is necessary to develop novel strategies to prevent cardiac IRI, thus improving the clinical outcome in patients with ischemic heart disease.
1. Introduction
2. Ferroptosis
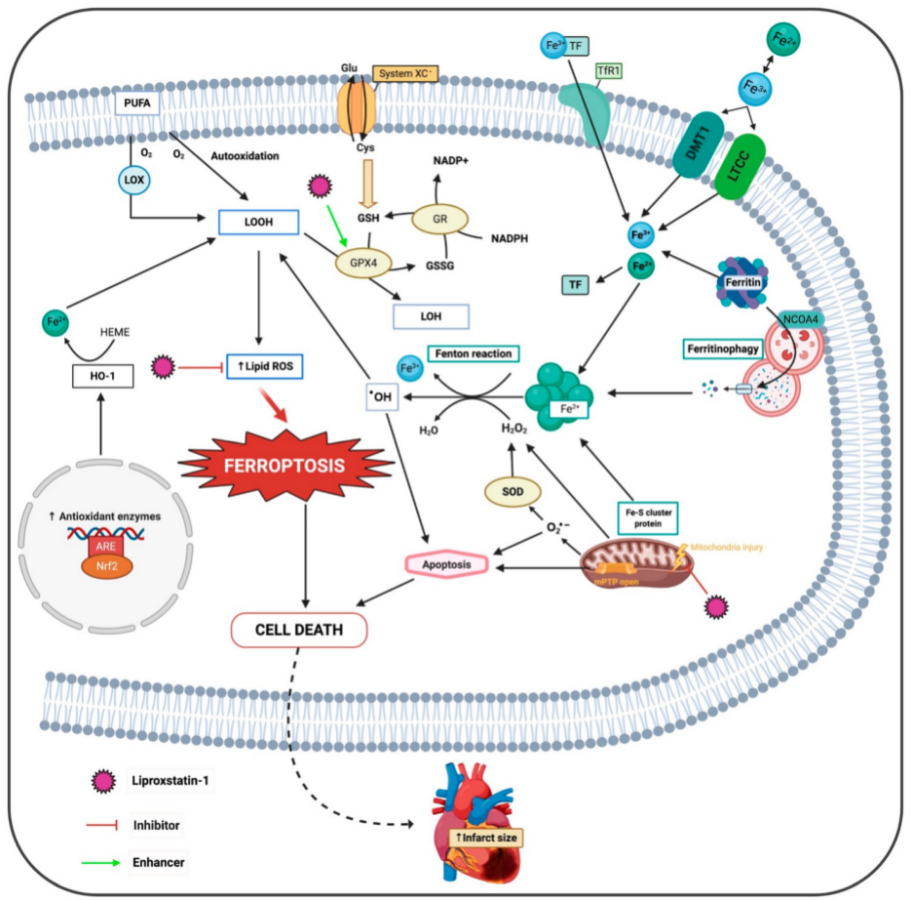
2.1. Class 1 Ferroptosis Inducers
2.2. Class 2 Ferroptosis Inducers
2.3. Lysosome and Ferroptosis
2.4. Ferroptosis and Relevance of the Cell Membrane
2.5. Autophagy-Induced Ferroptosis
2.6. Ferroptosis and Necroinflammation
2.7. Ferroptosis and Mitochondria in Cardiomyocytes
2.8. Cell Death Propagation and Ferroptosis
3. Does Ferroptosis Occur in the Ischemic Phase or Reperfusion Phase?
References
- World Health Organization. Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 14 May 2020).
- World Health Organization. The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 15 December 2020).
- Ruiz-Meana, M.; García-Dorado, D. Pathophysiology of Ischemia-Reperfusion Injury: New Therapeutic Options for Acute Myocardial Infarction. Rev. Española Cardiol. Engl. Ed. 2009, 62, 199–209.
- White, H.D.; Chew, D.P. Acute myocardial infarction. Lancet 2008, 372, 570–584.
- Garcia-Dorado, D.; Rodríguez-Sinovas, A.; Ruiz-Meana, M.; Inserte, J. Protection Against Myocardial Ischemia-reperfusion Injury in Clinical Practice. Rev. Española Cardiol. Engl. Ed. 2014, 67, 394–404.
- Yellon, D.M.; Hausenloy, D.J. Myocardial Reperfusion Injury. N. Engl. J. Med. 2007, 357, 1121–1135.
- Nakamura, T.; Naguro, I.; Ichijo, H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1398–1409.
- Li, W.; Feng, G.; Gauthier, J.M.; Lokshina, I.; Higashikubo, R.; Evans, S.; Liu, X.; Hassan, A.; Tanaka, S.; Cicka, M.; et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Investig. 2019, 129, 2293–2304.
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680.
- Gale, C.P.; Metcalfe, E.; West, R.M.; Das, R.; Kilcullen, N.; Morrell, C.; Crook, R.; Batin, P.D.; Hall, A.S.; Barth, J.H. An Assessment of the concentration-related prognostic value of cardiac troponin i following acute coronary syndrome. Am. J. Cardiol. 2011, 108, 1259–1265.
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191.
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234.
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972.
- Haeggström, J.Z.; Funk, C.D. Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem. Rev. 2011, 111, 5866–5896.
- Kagan, V.E.; Mao, G.; Qu, F.; Pedro, J.; Angeli, F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; et al. Oxidized Arachidonic/Adrenic Phosphatidylethanolamines Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90.
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St. Croix, C.M.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.e26.
- Sparvero, L.J.; Tian, H.; Amoscato, A.A.; Sun, W.-Y.; Anthonymuthu, T.S.; Tyurina, Y.Y.; Kapralov, O.; Javadov, S.; He, R.-R.; Watkins, S.C.; et al. Direct Mapping of Phospholipid Ferroptotic Death Signals in Cells and Tissues by Gas Cluster Ion Beam Secondary Ion Mass Spec-trometry (GCIB-SIMS). Angew. Chemie Int. 2021.
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209.
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072.
- Yagoda, N.; Von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868.
- Baba, Y.; Higa, J.K.; Shimada, B.K.; Horiuchi, K.M.; Suhara, T.; Kobayashi, M.; Woo, J.D.; Aoyagi, H.; Marh, K.S.; Kitaoka, H.; et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am. J. Physiol. Hear. Circ. Physiol. 2018, 314, 659–668.
- Fang, X.; Cai, Z.; Wang, H.; Han, D.; Cheng, Q.; Zhang, P.; Gao, F.; Yu, Y.; Song, Z.; Wu, Q.; et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ. Res. 2020, 127, 486–501.
- Kobayashi, M.; Suhara, T.; Baba, Y.; Kawasaki, N.K.; Higa, J.K.; Matsui, T. Pathological Roles of Iron in Cardiovascular Disease. Curr. Drug Targets 2018, 19, 1068–1076.
- Alu, A.; Han, X.; Ma, X.; Wu, M.; Wei, Y.; Wei, X. The role of lysosome in regulated necrosis. Acta Pharm. Sin. B 2020, 10, 1880–1903.
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379.
- Angeli, J.P.F.; Shah, R.; Pratt, D.A.; Conrad, M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol. Sci. 2017, 38, 489–498.
- Stamenkovic, A.; O’Hara, K.A.; Nelson, D.C.; Maddaford, T.G.; Edel, A.L.; Maddaford, G.; Dibrov, E.; Aghanoori, M.; Kirshenbaum, L.A.; Fernyhough, P.; et al. Oxidized phosphatidylcholines trigger ferroptosis in cardiomyocytes during ischemia/reperfusion injury. Am. J. Physiol. Circ. Physiol. 2021.
- Srikantan, S.; Gorospe, M. HuR function in disease. Front. Biosci. 2012, 17, 189–205.
- Chen, H.-Y.; Xiao, Z.-Z.; Ling, X.; Xu, R.-N.; Zhu, P.; Zheng, S.-Y. ELAVL1 is transcriptionally activated by FOXC1 and promotes ferroptosis in myocardial ischemia/reperfusion injury by regulating autophagy. Mol. Med. 2021, 27, 1–14.
- Zhou, T.; Chuang, C.C.; Zuo, L. Molecular Characterization of Reactive Oxygen Species in Myocardial Ischemia-Reperfusion Injury. BioMed Res. Int. 2015, 2015, 864946.
- Hannenhalli, S.; Kaestner, K.H. The evolution of Fox genes and their role in development and disease. Nat. Rev. Genet. 2009, 10, 233–240.
- Sarhan, M.; Land, W.G.; Tonnus, W.; Hugo, C.P.; Linkermann, A. Origin and consequences of necroinflammation. Physiol. Rev. 2018, 98, 727–780.
- Tait, S.W.G.; Ichim, G.; Green, D.R. Die another way-non-apoptotic mechanisms of cell death. J. Cell Sci. 2014, 127, 2135–2144.
- Proneth, B.; Conrad, M. Ferroptosis and necroinflammation, a yet poorly explored link. Cell Death Differ. 2019, 26, 14–24.
- Linkermann, A.; Stockwell, B.R.; Krautwald, S.; Anders, H.J. Regulated cell death and inflammation: An auto-amplification loop causes organ failure. Nat. Rev. Immunol. 2014, 14, 759–767.
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248.
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841.
- Martín-Fernández, B.; Gredilla, R. Mitochondria and oxidative stress in heart aging. Age 2016, 38, 225–238.
- Loor, G.; Kondapalli, J.; Iwase, H.; Chandel, N.S.; Waypa, G.B.; Guzy, R.D.; Vanden Hoek, T.L.; Schumacker, P.T. Mitochondrial oxidant stress triggers cell death in simulated ischemia-reperfusion. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1382–1394.
- Li, N.; Jiang, W.; Wang, W.; Xiong, R.; Wu, X.; Geng, Q. Ferroptosis and its emerging roles in cardiovascular diseases. Pharmacol. Res. 2021, 166, 105466.
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2020, 99, 151058.
- Škárka, L.; Ošťádal, B. Mitochondrial membrane potential in cardiac myocytes. Physiol. Res. 2002, 51, 425–434.
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3.
- Liang, H.; Van Remmen, H.; Frohlich, V.; Lechleiter, J.; Richardson, A.; Ran, Q. Gpx4 protects mitochondrial ATP generation against oxidative damage. Biochem. Biophys. Res. Commun. 2007, 356, 893–898.
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308.
- Griffith, O.W.; Meister, A. Origin and turnover of mitochondrial glutathione. Proc. Natl. Acad. Sci. USA 1985, 82, 4668–4672.
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700.
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151.
- Kim, S.E.; Zhang, L.; Ma, K.; Riegman, M.; Chen, F.; Ingold, I.; Conrad, M.; Turker, M.Z.; Gao, M.; Jiang, X.; et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat. Nanotechnol. 2016, 11, 977–985.
- Riegman, M.; Sagie, L.; Galed, C.; Levin, T.; Steinberg, N.; Dixon, S.J.; Wiesner, U.; Bradbury, M.S.; Niethammer, P.; Zaritsky, A.; et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat. Cell Biol. 2020, 22, 1042–1048.
- Riegman, M.; Bradbury, M.S.; Overholtzer, M. Population Dynamics in Cell Death: Mechanisms of Propagation. Trends Cancer 2019, 5, 558–568.
- Tang, L.J.; Luo, X.J.; Tu, H.; Chen, H.; Xiong, X.M.; Li, N.S.; Peng, J. Ferroptosis occurs in phase of reperfusion but not ischemia in rat heart following ischemia or ischemia/reperfusion. Naunyn. Schmiedebergs. Arch. Pharmacol. 2021, 394, 401–410.
- Feng, Y.; Madungwe, N.B.; Imam Aliagan, A.D.; Tombo, N.; Bopassa, J.C. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem. Biophys. Res. Commun. 2019, 520, 606–611.
- Berenshtein, E.; Vaisman, B.; Goldberg-Langerman, C.; Kitrossky, N.; Konijn, A.M.; Chevion, M. Roles of ferritin and iron in ischemic preconditioning of the heart. Mol. Cell. Biochem. 2002, 234–235, 283–292.
- Chevion, M.; Jiang, Y.; Har-El, R.; Berenshtein, E.; Uretzky, G.; Kitrossky, N. Copper and iron are mobilized following myocardial ischemia: Possible predictive criteria for tissue injury. Proc. Natl. Acad. Sci. USA 1993, 90, 1102–1106.
- Tang, W.H.; Wu, S.; Wong, T.M.; Chung, S.K.; Chung, S.S.M. Polyol pathway mediates iron-induced oxidative injury in ischemic-reperfused rat heart. Free Radic. Biol. Med. 2008, 45, 602–610.
- Chang, H.; Wu, R.; Shang, M.; Sato, T.; Chen, C.; Shapiro, J.S.; Liu, T.; Thakur, A.; Sawicki, K.T.; Prasad, S.V.; et al. Reduction in mitochondrial iron alleviates cardiac damage during injury. EMBO Mol. Med. 2016, 8, 247–267.
- Jian, Y.; Lesnefsky, E.J. Incremental iron overload during reperfusion progressively augments oxidative injury. Chin. Med. J. Engl. 1996, 109, 450–458.
- Heger, M.; Reiniers, M.J.; Van Golen, R.F. Mitochondrial metabolomics unravel the primordial trigger of ischemia/reperfusion injury. Gastroenterology 2015, 148, 1071–1073.
- Li, Y.; Feng, D.; Wang, Z.; Zhao, Y.; Sun, R.; Tian, D.; Liu, D.; Zhang, F.; Ning, S.; Yao, J.; et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019, 26, 2284–2299.

