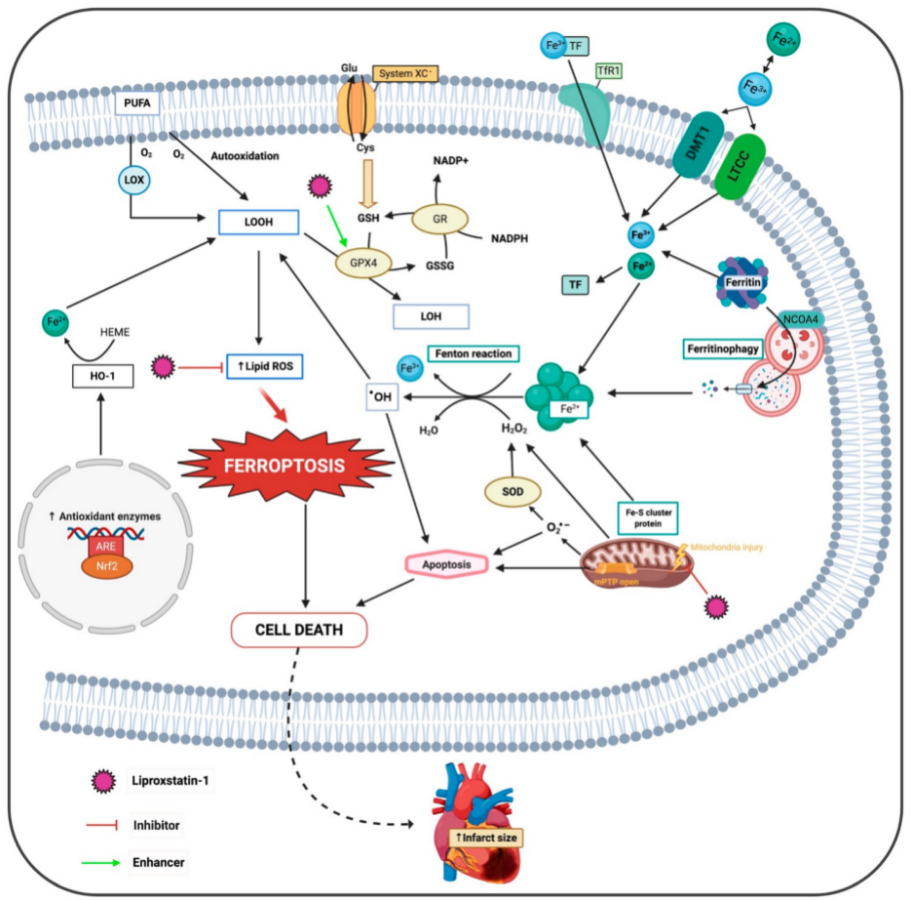
2. Ferroptosis
There are two main routes of cell death, apoptotic and non-apoptotic. Apoptosis is a highly controlled cell death process performed by healthy and damaged cells in response to a physiological or pathological stimulus, including I/R events
[10][69]. On the other hand, in non-apoptotic cell death
reswe
archers can highlight ferroptosis, a regulated forms of necrosis that is caused by the accumulation of lipid peroxidation products and ROS derived from iron metabolism, mainly when GSH levels in the cell are depleted or when glutathione peroxidase 4 (GPX4) enzyme is inhibited
[11][70]. GPX4 converts phospholipid hydroperoxides to lipid alcohols using reduced GSH, thus inhibiting ferroptosis
[12][71] (
Figure 1Figure 2).
Figure 1. Molecular mechanisms of the deleterious effects of ferroptosis and proposed sites of protective action of liproxstatin-1. ARE, antioxidant response elements; DMT1, divalent metal transporter; Fe2+, ferrous iron; Fe3+, ferric iron; GPX4, glutathione peroxidase 4; GR, glutathione reductase; HO-1, heme oxygenase 1; H2O2, hydrogen peroxide; LOH, lipid alcohols; LOOH, lipid hydroperoxides; LOX, lipoxygenase; LTCC, L-type calcium channel; mPTP, mitochondrial permeability transition pore; Nrf2, nuclear factor-erythroid 2-related factor 2; NCOA4, nuclear receptor coactivator 4; O2•−, superoxide radical; •OH, hydroxyl radical; PUFA, poly-unsaturated fatty acids; SOD, superoxide dismutase; TF, transferrin; TfR1, transferrin receptor.
Furthermore, lipid peroxidation occurs by two main mechanisms, an iron-catalyzed spontaneous peroxyl radical-mediated reaction called autoxidation and an enzyme-mediated process catalyzed by LOXs
[13][72]. LOXs are iron-containing enzymes that catalyze polyunsaturated fatty acid (PUFA) dioxygenation, thus producing lipid hydroperoxides that accumulate in RE
[14][15][73,74]. These enzymes, primarily 15-LOX, modulate ferroptosis by specifically oxidizing phosphatidylethanolamines (PE), with specificity towards two fatty acyls-arachidonoyl (FAA) and adrenoyl, generating OOH-PE species that act as ferroptotic signals
[15][74]. A recent study has identified a small scaffolding protein inhibitor of kinase cascades, phosphatidylethanolamine-binding protein 1, which complexes with two isoforms of 15-LOX, 15-LO1 and 15-LO2, and changes their substrate affinity from free FAA to FAA-PE to generate OOH-FAA-PE
[16][75]. Recently, new imaging technologies have permitted the detection and visualization of OOH-PE species in ferroptotic cardiomyocytes
[17][76].
Currently, there are two classes of ferroptosis inducers that target specific proteins in the ferroptotic pathway
[18][77], described below.
2.1. Class 1 Ferroptosis Inducers
Class 1 ferroptosis inducers such as erastin are one of the main inducers of ferroptosis, which blocks the XC-system, the cystine/glutamate exchanger of the membrane, blocking the entry of cystine into the cell, which is necessary for the synthesis of GSH
[19][78] (
Figure 1Figure 2. Erastin also binds to and inhibits voltage dependent anion channels (VDAC) 2 and VDAC 3, which also leads to cell death
[20][79]. Previous studies demonstrated that although there is no XC-system in the heart, cardiomyocyte death also occurs, but is due to erastin induction
[21][80]. Despite this, the XC-system is encoded by the gene Slc7a11 and its selective overexpression in cardiomyocytes increased GSH levels and prevented cardiac ferroptosis
[22][68].
2.2. Class 2 Ferroptosis Inducers
Class 2 ferroptosis inducers such as Ras Selective Lethal 3 directly inhibit GPX4, increase lipid peroxidation, and induce ferroptosis, thus triggering the accumulation of lipid ROS and resulting in cell death
[23][81].
2.3. Lysosome and Ferroptosis
There are a variety of organelles that interfere with iron metabolism and are associated with ferroptosis. Lysosome is part of this group because it can modulate iron equilibrium, causing a burst of ROS expression within it, which is attributable to its acid pH and high iron content. Lipid peroxidation may occur in the lysosome membrane due to ROS accumulation and iron overload. Consequently, the lysosomal membrane is permeabilized, causing a greater generation of radicals, cell membrane denaturation, and GSH consumption
[24][82].
Moreover, production of lipid ROS, mitochondrial shrinkage, increased mitochondrial membrane density, and involvement of important transcription factors to produce an antioxidant or pro oxidant response are the main features of ferroptosis. These characteristics make ferroptosis different from other nonapoptotic cell death programs such as apoptosis
[25][26][83,84]. Studies have revealed that ferroptosis is one of the major drivers of myocardial infarction
[9].
2.4. Ferroptosis and Relevance of the Cell Membrane
Cell membranes are composed of phosphatidylcholine (PC), which is one of the principal classes of phospholipids. A relevant feature of PC is that it is highly vulnerable to oxidation, forming species related to different pathologies. Oxidized phospholipids (OxPCs) induce cell death only through ferroptosis and not because of apoptosis or necrosis. OxPCs affect the functionality of cardiomyocytes such as changes in Ca
2+ transients and net cardiomyocyte contraction and can be responsible for reperfusion arrhythmias
[27][85].
2.5. Autophagy-Induced Ferroptosis
Ferroptosis is closely related to autophagy, with many molecules involved. Embryonic lethal-abnormal vision like protein1 (ELAVL1) is a protein coding gene that regulates gene expression by stabilizing message RNAs such as TNF-α or VEGF-A and is related to the process of cell death and oxidative stress
[28][86]. ELAVL1 inhibition decreases AMI inflammation responses, so it has a role in myocardial IRI, wherein excessive ROS and inflammatory cytokines were produced with a substantial increase of ELAVL1
[29][30][87,88]. Forkhead box C1 (FOXC1) is a transcription factor that has a role in cell growth and survival and also in heart diseases
[31][89].
A recent study
[29][87] showed that FOXC1 transcriptionally-activated ELAVL1 strongly contributes to myocardial IRI through an increase in the autophagic ferroptosis pathway. During I/R, increased ELAVL1 expression produces a deleterious effect on enzyme function and cellular antioxidant capacity because both GSH and GPX4 levels decrease. Nevertheless, ELAVL1 inhibition suppresses ferroptosis and myocardial IRI, restoring GPX4 levels and recovering the viability of myocardial cells, thereby reducing cell injury. The greatest anti-ferroptotic effect of knockdown ELAVL1 is that it inhibits I/R-induced autophagy, thereby protecting against ferroptosis, heart IRI and reducing myocardial infarct size. Moreover, ELAVL1 levels decrease if FOXC1 is knocked down, demonstrating that FOXC1 regulates ELAVL1 expression during I/R. Therefore, the sequence is that ELAVL1 knockdown inhibits ferroptosis and heart IRI and also inhibits autophagy. Autophagy-dependent ferroptosis undoes the effects of ELAVL1 knockdown and contributes to heart IRI, producing overproduction of lipid signaling. This relation between FOXC1 and ELAVL1 and their link to ferroptosis can be a beneficial target against myocardial IRI
[29][87].
2.6. Ferroptosis and Necroinflammation
Normally, necrosis is strongly associated with a pro-inflammatory response, where necroinflammation is the immune response to necrosis in a living organism. This can be through an unregulated form such as traumatic necrosis or signaling pathways defined as necroptosis, ferroptosis and pyroptosis, executing necrosis as a regulated process
[32][90]. This cell death pathway, unlike other cell death pathways such as apoptosis, does not present a silent inflammatory response
[33][91]. The evidence of necroinflammation and its relationship with ferroptosis is very poor and much remains to be investigated
[34][92] but recent studies show that ferroptotic cells collapse and release molecular mediators associated with pro-inflammatory damage
[35][93]. Cell markers of the immune system can be detected in various tissues and suggest that ferroptotic cells release immune-stimulating cellular components
[36][94]. So far, most studies in pathologies and genetic models of ferroptosis in vivo have focused on two specific organs: kidney and brain. There is evidence that necroinflammation can occur in the ferroptotic tissue of the kidney, with infiltration of neutrophils and activation of macrophages
[37][95].
RWe
searchers know that IRI in the heart is associated with an inflammatory response and
researcherswe hypothesize that in the heart tissue, due to the infiltration of inflammatory molecules, a response similar to the kidneys will occur, modulating the final size of the AMI. Hence, it is also necessary to advance studies of ferroptosis in humans.
2.7. Ferroptosis and Mitochondria in Cardiomyocytes
The heart has an aerobic metabolism and is dependent on mitochondrial function to obtain energy because cardiomyocytes consume more than 90% of intracellular ATP
[38][96]. For this reason, mitochondria have been studied as a promising target that could be involved in ferroptosis.
During I/R, there is an increase in the oxidation of the mitochondrial matrix and many morphological and metabolic changes occur within the mitochondria.
RWe
searchers can highlight rounding and blebbing of the mitochondrial outer membrane. Disorganization and dispersion are also evident
[39][97]. Other morphological changes include a reduction of mitochondria size with condensed mitochondrial membrane densities, reduction of mitochondria cristae, and outer mitochondrial membrane rupture
[40][98]. Some of the changes in the morphological features of mitochondria are not related to other cell death processes, such as necrosis, apoptosis or autophagy, highlighting the relevance of ferroptosis
[41][99].
In fact, there is no consensus on the role of mitochondria in ferroptosis. Nevertheless, recent evidence has demonstrated that mitochondria are organelles involved in the execution of different mechanisms of cell death including ferroptosis, specifically by opening of mPTP and alteration of mitochondrial outer membrane permeabilization
[41][99]. The mitochondrial membrane potential (MMP) is commonly used to evaluate the mitochondrial function, where a loss of the MMP means mitochondrial dysfunction
[9][42][9,100] and maintaining the MMP is very important to the survival and function of cells that require high-energy, such as myocardiocytes
[42][100].
During mitochondrial-mediated apoptosis, a decrease in MMP was observed before final cell death. This decrease in potential indicates increased permeability of the mitochondrial outer membrane, an important feature of mitochondrial-mediated apoptosis
[43][101]. Although the release of cytochrome C during reperfusion leads to the activation of caspases, interventions exist that inhibit BH3-dependent apoptosis, but do not protect against cell death. This indicates that although apoptosis is activated in I/R, it is not necessary for subsequent cell death, suggesting that other mechanisms may exist to produce cell death
[39][97] such as ferroptosis. Indeed, the role of the mitochondria in this type of cell death is much more proactive than in apoptosis
[43][101].
Gao et al. showed that both mitochondrial tricarboxylic acid (TCA) cycle and mETC play a crucial role in cysteine-deprivation-induced ferroptosis. TCA function supports mETC activity by regulating different complexes in the inner membrane of mitochondria and mETC components are necessary to allow accumulation of lipid ROS, inducing ferroptosis. Indeed, inhibitors of mitochondrial complexes suppressed lipid ROS accumulation and ferroptosis. Moreover, glutaminolysis is associated with I/R injury through ferroptosis and also provides several intermediates into the mitochondrial TCA cycle. Glutamine metabolism has been described as a key event in ferroptosis induction. However, a number of TCA cycle metabolites such as alpha-ketoglutaric acid, succinate, fumarate and malate can recapitulate the function of glutamine in lipid ROS accumulation and ferroptosis induced by cysteine deprivation or XC-system inhibition, indicating that the TCA cycle is required for ferroptosis
[43][101]. It is a known fact that the mitochondria produce ATP using the potential electrochemical proton gradient across the mitochondrial membrane, which is the result of the action of the TCA cycle and mETC. In this sense, it has been described that glutaminolysis, the TCA cycle, and other ferroptosis inducers lead to MMP hyperpolarization, which is associated with ferroptosis and, eventually, may cause the membrane to collapse. Presumably, this hyperpolarization reflects an increase in mETC activity and the subsequent generation and accumulation of lipid ROS
[43][101], where the mitochondrial outer membrane has many PUFAs so the MMP is very sensitive to lipid peroxidation
[44][102]. Thus, glutaminolysis is an essential factor for ferroptosis and its inhibition can protect heart tissue from IRI. Indeed, glutaminolysis inhibitors recover MMP, reduce myocardial infarct size and improve the heart’s function
[43][45][101,103]. In addition, metabolic changes during I/R initially include a decrease in mitochondrial respiration and an increase in glycolysis. Then, during reperfusion, there is an increase in mitochondrial respiration due to increased oxygen and, consequently, ROS explosion and execution of ferroptosis. The explosion could be due to increased metabolism of the mitochondria because more metabolites are entering
[41][99].
Although mitochondrial function is important, when mETC and glutaminolysis are blocked, ferroptosis will still occur if GPX4 is inhibited. Therefore, it follows that GPX4 has a more direct and effective effect than the other pathways. GPX4 overexpression allows better ATP production, maintains MMP and protects the heart against oxidative damage
[44][102]. In addition, erastin-induced ferroptosis does not require ETC to induce ferroptosis
[19][78]. This indicates that mitochondrial function is dispensable for ferroptosis. Regarding this point, it is important to note that the role of the mitochondria depends on the context. On the one hand, blocking mitochondrial function potently inhibits ferroptosis. On the other hand, GPX4 elimination triggers ferroptosis in cells independent of mitochondria
[43][101]. A recent study demonstrated that OxPCs decrease GPX4 levels and have a direct effect on the mitochondria. During I/R, the release of OxPCs can decrease mitochondrial spare respiratory capacity and, because of this, the ability of mitochondria to respond against oxidative stress is decreased, producing less ATP and, as a consequence, cell death via ferroptosis
[27][85].
Apart from glutaminolysis and the TCA cycle, GSH also plays an important role in the mitochondria. This organelle does not have the CAT enzyme to act on O2
•− or the enzymes required for GSH synthesis. GSH acts non enzymatically but is also an essential cofactor for GSH-linked antioxidant enzymes such as GPXs. Under oxidative stress, oxidized glutathione levels increase and the relative proportion between the reduced and the oxidized forms (GSH/GSSG) is considered a marker of oxidative stress. Mitochondrial GSH (mGSH) levels are similar to GSH levels in the cytosol
[46][104]. In terms of mitochondrial oxidative stress balance, mGSH is a critical factor in the control of cell survival/death and regulates factors that influence MMP permeability. Its depletion induces cellular damage and promotes cell death
[47][48][105,106].
Previously,
reswe
archers identified glutamine as a serological component that is involved in ferroptosis and mitochondria. However, there is another component that is also found in high amounts in the blood, which is TF. This intracellular pathway, including its membrane receptor, is required for ferroptosis. Although this unexpected role of TF is not entirely clear, it is believed that due to its high blood level, a partial cysteine deprivation is sufficient to produce necrosis. Furthermore, under pathological conditions such as I/R, cells are more susceptible to ferroptosis
[45][103].
In summary, ferroptosis can be triggered by different biological pathways. This can be caused by both intracellular and extracellular mechanisms and there are conditions that cause the cell to be more susceptible to this type of cell death such as cysteine deprivation or low levels of GSH
[45][103].
2.8. Cell Death Propagation and Ferroptosis
It has been described that ferroptosis importantly involves intercellular interaction, where cell death is transmitted to neighboring cells, spreading in a wave-like manner
[37][49][95,107], which is characteristic of particular forms of ferroptosis
[50][108], while apoptosis is generally classified as an autonomous cell death that does not induce death in the other cells around it
[51][109].
A recent study describes two different types of ferroptosis based on the spread of cell death. On one hand is cell-autonomous ferroptosis, occurring when GPX4 is inhibited, and on the other is propagative ferroptosis, which is induced by inhibiting the generation of GSH or increasing the concentration of iron
[50][108]. This study also demonstrated that iron and lipid peroxidation are necessary to the propagation of ferroptosis, but not for the cell rupture. However, the speed of the wave propagation is dependent on the lysis of the cell and the mechanism of plasma membrane permeabilization and propagation of cell death must be elucidated
[50][108].
Therefore,
reswe
archers hypothesize that this process could be one of the mechanisms that contribute more to the increase of the infarct size during reperfusion, considering that ferroptosis occurs in heart IRI
[9][45][52][53][9,103,110,111] together with an increase in the concentration of intracellular iron and lipid peroxidation
[9][25][54][55][56][57][58][9,61,62,67,83,112,113]. Therefore, it is necessary to conduct studies focused on heart IRI that can verify the appearance of these waves of ferroptosis in cardiac tissue exposed to I/R, along with its mechanism and importance in the damage process and thus reinforce the idea that ferroptosis is the most important driver of the final infarct size
[8][9][8,9].
3. Does Ferroptosis Occur in the Ischemic Phase or Reperfusion Phase?
Usually, the ischemic phase is overlooked because of the traditional view that exists that ischemia-induced tissue injury and loss of function is an arbitrary consequence of oxygen deprivation, so I/R studies have focused on the reperfusion phase but are of limited translational value
[59][60][114,115]. Two well recognized biomarkers for ferroptosis are GPX4 and long-chain-fatty-acid-CoA ligase 4 (ACSL4). When ACSL4 and GPX4 proteins levels were studied on ischemic heart, there were no significant changes during different points of ischemia, in addition to no significant changes in iron or MDA contents in the cardiac tissues. As the reperfusion time was extended, levels of ACSL4 protein were gradually elevated, concomitant with a decrease in GPX4 levels in the cardiac tissue, where a gradual increase in iron concentration was also obtained along with increased MDA levels in the myocardium. Therefore, th
eis study finally suggested that ferroptosis occurs during the reperfusion phase in rat hearts that are subjected to an I/R process and not during the ischemic phase
[52][110].
Consistently, these results not only appear in hearts, but also in the intestine and other organs. A previous study
[60][115] suggested that ferroptosis occurs at the early stage of reperfusion. Expression of ACSL4 was induced in ischemic intestines and GPX4 levels were reduced after 45 min of ischemia. These results may sensitize the intestine to ferroptosis reperfusion because this second phase was likely to lead to ferroptosis after 45 min of ischemia. After 15 min of reperfusion, rupture of the outer mitochondrial membrane occurs, and after 30 min the disappearance of mitochondrial cristae was more evident. Moreover, the expression of GPX4 was decreased at 30 min of reperfusion, whereas the expression of cyclooxygenase-2 was increased, as well as 12- (12-HETE) and 15- hydroxyeicosatetraenoic acid (15-HETE), both derived from arachidonic acid. The results showed that ferroptosis was more active 30 min after reperfusion and not during other moments of this phase
[60][115]. Moreover, the polyol pathway and ELAVL1 expression was remarkably elevated in reperfusion
[29][56][67,87] and in this phase, lysosomes are ready to be released
[24][82]. Therefore, these previous studies finally suggest that ferroptosis occurs during the reperfusion phase in IRI models and not during the ischemic phase, which is very important to take into consideration when developing cardioprotective therapies that inhibit ferroptosis to reduce heart IRI
[52][110].