Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Germana Falcone | -- | 2915 | 2022-05-02 09:17:16 | | | |
| 2 | Vivi Li | Meta information modification | 2915 | 2022-05-05 04:39:03 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Falcone, G.; , .; Battistini, J.I.; Provenzano, C.; Martelli, F.; Cardinali, B. Molecular Therapies for Myotonic Dystrophy Type 1. Encyclopedia. Available online: https://encyclopedia.pub/entry/22568 (accessed on 27 July 2026).
Falcone G, , Battistini JI, Provenzano C, Martelli F, Cardinali B. Molecular Therapies for Myotonic Dystrophy Type 1. Encyclopedia. Available at: https://encyclopedia.pub/entry/22568. Accessed July 27, 2026.
Falcone, Germana, , Jonathan Isacco Battistini, Claudia Provenzano, Fabio Martelli, Beatrice Cardinali. "Molecular Therapies for Myotonic Dystrophy Type 1" Encyclopedia, https://encyclopedia.pub/entry/22568 (accessed July 27, 2026).
Falcone, G., , ., Battistini, J.I., Provenzano, C., Martelli, F., & Cardinali, B. (2022, May 02). Molecular Therapies for Myotonic Dystrophy Type 1. In Encyclopedia. https://encyclopedia.pub/entry/22568
Falcone, Germana, et al. "Molecular Therapies for Myotonic Dystrophy Type 1." Encyclopedia. Web. 02 May, 2022.
Copy Citation
Myotonic dystrophy type 1 (DM1) is the most common muscular dystrophy affecting many different body tissues, predominantly skeletal and cardiac muscles and the central nervous system. The expansion of CTG repeats in the DM1 protein-kinase (DMPK) gene is the genetic cause of the disease. The pathogenetic mechanisms are mainly mediated by the production of a toxic expanded CUG transcript from the DMPK gene. With the availability of new knowledge, disease models, and technical tools, much progress has been made in the discovery of altered pathways and in the potential of therapeutic intervention, making the path to the clinic a closer reality.
myotonic dystrophy
trinucleotide-expansion disease
DM1 mice
antisense oligonucleotides
molecular therapy
gene editing
1. Introduction
Myotonic dystrophy type 1 (DM1) is caused by an unstable expanded CTG repeat located within the 3′ untranslated region (3′ UTR) of the DMPK gene. The molecular mechanisms of DM1 are mainly the consequence of accumulation of mutant DMPK transcripts into ribonuclear foci leading to the impairment of alternative splicing and normal gene expression. Cell and animal models of DM1 have been crucial to providing insight into disease mechanisms and to revealing new therapeutic targets.
2. Myotonic Dystrophy Type 1: Clinical Features and Pathogenetic Mechanisms
DM1 is the most common dystrophy in adults, having an estimated worldwide prevalence of 1:20,000, with a recent report of 4.76:10,000 for DMPK CTG expansion ≥50 CTG repeats in a newborn screening program in New York State, USA [1]. Clinical features of DM1 (Steinert’s disease, OMIM# 160900) include muscle weakness, dysphagia, neuromuscular respiratory insufficiency, cardiac complications and cognitive, intellectual or behavioral impairment as well as sleep disorders. In the most severe forms, life quality and expectancy are seriously compromised [2][3][4]. DM1 results from CTG-repeat expansions in the 3′ UTR of the DMPK gene on chromosome 19 [5][6]. The disease severity and age of onset are broadly correlated with the number of CTG repeats, with the highest (over 750) in the congenital form, while in non-affected individuals the number of repeats is up to 35 [2]. The number of repeats is usually unstable and tends to increase in some body tissues during lifetime (somatic instability) as well as in successive generations, leading to the phenomenon called “anticipation”, where children of DM1 patients have a higher repeat number and more severe phenotypes compared to their parents [7]. Interestingly, in DM1 families with variant repeats, where GGC, CCG and CTC interruptions are present within the CTG-repeat array, the repeats are stabilized and the disease phenotypes are milder [8][9]. Several pathogenic mechanisms likely contribute to disease in DM1 (Figure 1) [4][10]. At the DNA level, epigenetic modifications may impact the development or severity of the phenotype in DM1 patients [11]. In DM1 patient-derived cells and in a DM1 mouse model, the hairpin-like structures of the repeats can induce chromatin changes, such as CpG methylation, resulting in haploinsufficiency of DMPK and neighboring genes, or cause replication-fork stalling during DNA duplication, leading to cell stress [12][13][14]. Large experimental evidence supports the hypothesis of an RNA gain-of-function mechanism of the mutated DMPK transcript. CUG-containing RNAs sequester crucial nuclear factors of the muscleblind-like (MBNL) family into ribonuclear foci, thus preventing their normal functions that are mainly associated with the regulation of alternative splicing [15]. Splicing regulation is required for the proper development and maintenance of tissues in which the DMPK gene is highly expressed, such as in muscle and the nervous system [16]. The MBNL family and the CUGBP Elav-like family (CELF) are among the most important splicing regulators in skeletal and cardiac muscle, and act antagonistically on several pre-mRNA targets [17][18]. Nuclear retention of MBNL proteins in nuclear foci prevents pre-mRNA processing and export to the cytoplasm, leading to a decrease in protein translation, and the loss of functional MBNL1 is accompanied by CELF1 upregulation [19]. The increase in CELF1 levels is induced by protein-kinase-C (PKC)-mediated hyperphosphorylation, leading to protein stabilization [20]. Both sense and antisense repeated RNAs have been shown to contribute to the clinical phenotype of nucleotide-expansion diseases [21]. An antisense transcript emanating from the DMPK-adjacent SIX5 regulatory region spanning the CTG expansion was first identified in DM1 patient-derived cells. The transcript was shown to be converted into siRNAs, which are able to recruit DNA and histone methyltransferases, leading to heterochromatin formation [22]. Interestingly, in transgenic mice carrying the human DMPK locus, in addition to CUG-containing transcripts, CAG-containing transcripts were also found to form distinct ribonuclear foci [23].
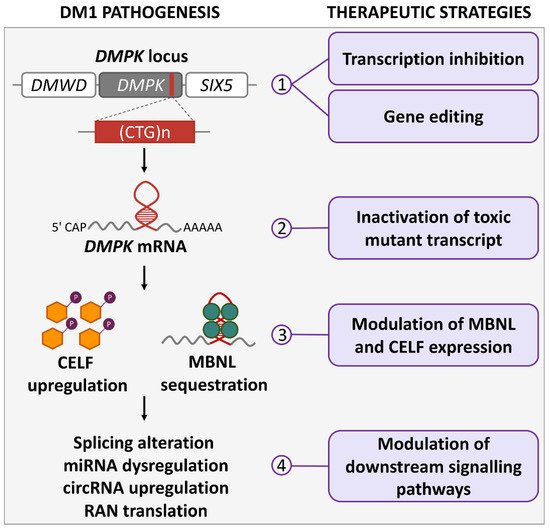
Figure 1. DM1 pathogenetic mechanisms and therapeutic strategies. The actions of molecular therapies for DM1 at different pathogenetic levels are illustrated: (1) at DMPK gene, drugs can inhibit CTG-repeat transcription and induce repeat contraction; ZFN, TALEN or CRISPR/Cas9 nucleases can modify gene sequence by inducing CTG-repeat contractions or deletions, or by inserting premature polyadenylation signals; (2) mutated DMPK mRNA can be functionally inactivated by drugs inducing degradation or binding to CUG repeats; (3) MBNL can be released from CUG repeats by disruption of MBNL:CUG interaction through competitive binding, and CELF levels can be regulated by protein kinase C and glycogen synthase kinase 3β; (4) altered signaling pathways downstream of DMPK transcript can be rescued by modulation of splicing and miRNAs; circRNAs and RAN translation could also be targets of future therapies.
Other mechanisms involved in DM1 pathogenesis are repeat-associated non-ATG (RAN) translation (reviewed in [24]), which results in the production of toxic protein aggregates containing polyglutamine from antisense CAG-repeated transcripts [25][26], microRNA (miRNA) dysregulation [27][28][29][30], and upregulation of circular RNA (circRNA) expression [31][32][33]. In addition to the ones described above, other signaling cascades are affected by the toxic DMPK RNA and may play important roles in DM1 pathogenesis. For example, MBNL and CELF regulators, besides being key splicing regulators, are likely involved in cytoplasmic pathogenetic processes altering proteostasis and sarcomere structure (reviewed in [34]). Omics studies have added new information to the previous knowledge, revealing several alterations in gene expression, alternative splicing, CpG methylation and proteins levels that potentially contribute to DM1 pathogenesis. In perspective, these new approaches can be crucial to evaluate the degree of therapeutic rescue and the off-target effects of drug candidates (reviewed in [35]).
3. DM1 Cell and Animal Models
In vitro models of DM1 have greatly contributed to clarifying the pathogenetic mechanisms of the disease. Among these, there are engineered cell lines with CTG repeats of different lengths inserted in minigenes [36][37], DM patient-derived primary myoblasts, immortalized myoblasts or MYOD1-converted fibroblasts [38][39][40][41], and embryonic stem cells [42]. Additionally, induced pluripotent stem cells (iPSCs) and iPSC-derived distinct cell types were used to study tissue-specific DM1 pathological alterations [43][44][45][46]. Recently, the first 3D in vitro human-muscle model of DM1 was developed by encapsulating patient-derived MYOD1-converted fibroblasts in hydrogels [47]. All of these cell models reproduce molecular alterations typical of DM1 and have been very useful for discovering crucial molecules and cellular pathways involved in the disease and for testing therapeutic strategies.
Drosophila models have also been used by several groups to study DM1. Interrupted CTG repeats of various lengths driven by either constitutive or inducible promoters were expressed in flies and DM1-related molecular as well as phenotypic alterations were observed in flies carrying more than 480 repeats [48][49][50]. Although DM1 modeling is complicated by the multifaceted impact of the DM1 mutation, many DM1 mouse models have been generated over time through the silencing of the Dmpk gene or the Mbnl family genes; alternatively, mouse models expressing CELF proteins or toxic CTG repeats in various tissues were produced, in order to mimic the different aspects of the disease and to discover therapeutic molecules (Table 1). It is unclear whether the DMPK haploinsufficiency observed in DM1 patients may affect functions of the tissues in which the gene is normally highly expressed, such as muscles and the central nervous system (CNS). To address this question, different Dmpk-knockout (KO) mouse models have been generated and characterized through the years with different results. Initial reports on Dmpk-KO mice described cardiac conduction defects [51] and mild myopathy [52]. Since these mice models were characterized by a mixed genetic background possibly leading to confounding effects, more recently a Dmpk-KO model backcrossed to two different pure genetic backgrounds was generated. This model did not confirm the previous observations, but showed that Dmpk gene deletion does not compromise cardiac or skeletal-muscle function [53]. Additionally, DMPK transcript silencing through antisense oligonucleotides (ASOs) was well tolerated in mice, rats and monkeys [54]. These findings suggest that reduction in DMPK expression should not be a prominent cause of the disease. Given the crucial regulatory role of MBNL proteins in DM1, different Mbnl KO models were generated to elucidate the role of each MBNL protein in the disease. Mbnl1 and Mbnl2 loss of function resulted in muscular and CNS symptom manifestation, respectively [55][56][57], while Mbnl3 KO caused a progressive delay in muscle regeneration and embryonic muscle differentiation abnormalities, in agreement with their expression profiles during development [58]. Mice with double Mbnl1/Mbnl2 KO or Mbnl1/Mbnl3 KO exhibited more severe phenotypes compared with the single KO [59][60] and the triple Mbnl1/2/3 KO in muscle tissues recapitulated the severe phenotype observed in congenital DM1, in both newborn and adult mice [61], supporting the idea of a prominent role of MBNL proteins and alternative splicing dysregulation in DM1 pathogenesis. To determine the role of CELF proteins in DM1 pathogenesis, mouse models overexpressing CELF1 and CELF2 in skeletal and/or cardiac muscle were generated [62][63][64][65]. Overexpression of CELF1 was shown to reproduce DM1-associated histopathological and functional changes [63]. Notably, CELF1/2 overexpressing mouse models have revealed a strong pattern of antagonistic regulation of mRNA levels by CELF and MBNL proteins through competitive binding to 3′ UTR regions [64] (Table 1A).
Table 1. DM1 mouse models used for studying pathogenetic mechanisms and/or molecular therapies.
| (A) Knockout and Overexpressing Models | ||||||||||||
| Mouse Model | Generation Strategy | DM1-Like Features | Limitations | Research Application | Ref | |||||||
| DMPK-/- | Dmpk KO via replacement of 5′-UTR and exons 1-7 with hygromycin cassette | Late-onset mild myopathy and altered Ca++ homeostasis | Mild phenotype; possible confounding insertional effects on flanking genes; mixed genetic background | Relevance of absence of DMPK protein to DM1 phenotype | [52][66] | |||||||
| DMPK-/- | Dmpk KO via replacement of 5′-UTR and exons 1-7 with neomycin cassette | Late-onset mild myopathy; decreased force generation; altered Na+ currents in skeletal muscles; cardiac conduction defects | Mild phenotype; possible confounding insertional effects on flanking genes; mixed genetic background | Relevance of absence of DMPK protein to DM1 phenotype | [51][67] | |||||||
| DMPK-/- | Dmpk KO via replacement of 5′-UTR and exons 1-7 with hygromycin cassette | No phenotype | Failure to replicate the DM1 phenotype | Relevance of absence of DMPK protein to DM1 phenotype | [53] | |||||||
| Mbnl1ΔE3/ΔE3 | Mbnl1 KO via targeted deletion of Mbnl1 exon 3 | Mild myotonia and myopathy (centralized nuclei, split fibers); heart conduction defects; progressive cataracts; AS alterations | Mild muscle phenotype; mild brain alterations; limited spliceopathy | Evaluation of MBNL1 splicing regulation to DM1 phenotype | [56][57] | |||||||
| Mbnl2ΔE2/ΔE2 | Mbnl2 KO via targeted deletion of Mbnl2 exon 2 | Development of several CNS alterations (REM sleep propensity, deficit in spatial memory, decreased synaptic plasticity), AS alterations | Failure to replicate the DM1 muscular phenotype | Evaluation of MBNL2 splicing regulation to DM1 phenotype | [55] | |||||||
| Mbnl3ΔE2 | Mbnl3 KO via targeted deletion of Mbnl3 exon 2 (X-linked) | Progressive delay in muscle regeneration; abnormalities in embryonic muscle differentiation leading to neonatal hypotonia | Possible compensation by MBNL3 truncated isoform or other MBNl family members | Evaluation of MBNL3 contribution to DM1 phenotype | [58] | |||||||
| Mbnl1ΔE3/ΔE3; Mbnl2C/C; Myo-Cre+/- |
Mbnl1 KO; skeletal-muscle specific Cre-mediated Mbnl2 KO | Small size at birth and skeletal abnormalities; myopathy and severe motor deficits; AS alterations also in brain tissues | High neonatal mortality and reduced lifespan | Evaluation of MBNL1 and MBNL2 contribution to DM1 muscular phenotype | [60] | |||||||
| Mbnl1ΔE3/ΔE3; Mbnl3ΔE2 |
Mbnl1 and Mbnl3 KO via targeted deletion of Mbnl1 exon 3 and Mbnl3 exon 2 | Myotonia and myopathy; reduction in muscle strength; chloride currents alteration; AS alterations; translation defects | AS alterations similar to Mbnl1 single knock out; lack of brain alterations | Evaluation of MBNL1 and MBNL3 contribution to DM1 phenotype | [59] | |||||||
| Mbnl1ΔE3/ΔE3; Mbnl2C/C; Mbnl3C; Myo-Cre+/- |
Mbnl1 KO; muscle-specific Cre-mediated Mbnl2 and Mbnl3 KO | Severe congenital myopathy and spliceopathy, severe respiratory difficulties and muscle wasting in adults; gene expression changes | High neonatal mortality and reduced lifespan | Evaluation of all MBNL proteins loss contribution to DM1 muscular phenotype | [61] | |||||||
| MCKCUGBP1 | Insertion of human CELF1 transgene under striated-muscle-specific MCK mouse promoter | Chains of central nuclei in myofibers, increased NADH reactivity, degenerating fibers and AS alterations | Neonatal lethality in mice expressing high levels of CELF1 | Contribution of CELF1 overexpression to DM1 muscular phenotype | [62] | |||||||
| TRECUGBP1 | Insertion of Tet-responsive human CELF1 transgene; heart-specific rtTA expression | Left ventricular systolic dysfunction and dilatation, AS alterations | DM1-like phenotype limited to heart defects | Contribution of CELF1 overexpression to DM1 heart phenotype | [63] | |||||||
| TRECUGBP1 | Insertion of Tet-responsive human CELF1 transgene; skeletal-muscle-specific rtTA expression | Myofibers containing central nuclei, decreased muscle weight, impaired muscle function, AS alterations | DM1-like phenotype limited to skeletal-muscle defects | Contribution of CELF1 overexpression to DM1 skeletal-muscle phenotype | [65] | |||||||
| TRECUGBP2 | Insertion of Tet-responsive human CELF2 transgene; heart-specific rtTA expression | No observed heart pathology; AS alterations similar to those observed in TRECUBP1 mice | Mild heart phenotype | Contribution of CELF2 overexpression to DM1 heart phenotype | [64] | |||||||
| (B) Transgenic Models with Repeat Expansion | ||||||||||||
| Mouse Model | Generation Strategy | (CTG)n | DM1-Like Features | Limitations | Research Application | Ref | ||||||
| DM200 | Insertion of a Tet-responsive expanded DMPK transgene where DMPK coding region is replaced by GFP | 200 | Ribonuclear foci; MBNL1 sequestration; AS alterations; myotonia, progressive cardiac conduction abnormalities | Splicing alterations in the heart have not been described | Study of DM1 phenotype associated with toxic CUG repeats; modeling muscle regeneration; test of therapeutic strategies | [68][69][70] | ||||||
| DM300 | Insertion of a 45Kb human genomic fragment containing DMWD, DMPK and SIX5 genes from a DM1 patient | ~300 | Ribonuclear foci (skeletal muscle, heart and brain); myotonia; muscle atrophy; morphological abnormalities; changes in the distribution of MAPT/Tau protein isoform; defect in glucose metabolism | High mortality; mild splicing alterations; intergenerational instability of CTG-repeat numbers | Evaluation of DMPK transcript toxicity in different tissues | [71][72] | ||||||
| DMSXL | Insertion of a 45Kb human genomic fragment containing DMWD, DMPK and SIX5 genes from a DM1 patient | >1000 | Ribonuclear foci; MBNL1 sequestration; AS alterations; deficits in motor performance; behavioral abnormalities; synaptic dysfunction; inhibition of exploratory activity and cerebellar glial dysfunction | High mortality; severe body-weight reduction; interindividual variability; decreased transgene expression with aging; mild muscular phenotype | Evaluation of DMPK transcript toxicity in different tissues and in multiple brain cell types; test of therapeutic strategies | [23][73][74] | ||||||
| HSALR | Insertion of the human skeletal actin (HSA) gene including CTG repeats in the 3’ UTR | ~250 | Ribonuclear foci; AS alterations; myotonia and muscle histopathology abnormalities (increase in central nuclei and variability in fiber size) after six months of age |
Limited to skeletal muscle; does not contain DMPK gene sequence; absence of muscle weakness | Investigation of expanded-CUG-repeat toxicity in muscle fibers; test of therapeutic strategies |
[75][76] | ||||||
| LC15 | Insertion of CTG expanded DMPK 3’ UTR downstream Luciferase gene driven by CMV-βA promoter | 250–400 | Ribonuclear foci, AS alteration and MBNL2 upregulation in the heart; reduced Na+ and K+ channel activity; ventricular arrhythmias | DM1-like phenotype limited to heart defects | Evaluation of biophysical mechanisms reproducing DM1-like electrocardiograph abnormalities | [77] | ||||||
| EpA960/ 𝛼-MHC-Cre |
Insertion of CTG expanded DMPK exon 15 transgene containing Cre-responsive loxP sequences; heart-specific myosin Cre expression | 960 (CTCGA-interrupted) |
Ribonuclear foci; MBNL1 sequestration; CELF1 protein upregulation; AS alterations; cardiomyopathy, arrhythmias; systolic and diastolic dysfunction |
Does not reproduce CTG-repeat continuity; mouse model no longer available | Evaluation of DMPK transcript toxicity and CELF1 overexpression in heart tissue | [78] | ||||||
| EpA960/ HSA-Cre |
Insertion of CTG expanded DMPK exon 15 transgene containing Cre-responsive loxP sequence; skeletal-muscle-specific Cre expression | 960 (CTCGA-interrupted) |
Ribonuclear foci; MBNL1 sequestration; CELF1 protein upregulation; AS defects; myotonia and progressive muscle wasting, deficits in muscle performance and histopathological abnormalities | Does not reproduce CTG-repeat continuity; mouse model no longer available | Evaluation of DMPK transcript toxicity and CELF1 overexpression in skeletal tissue | [79] | ||||||
| EpA960/ CamKII-Cre |
Insertion of CTG expanded DMPK exon 15 transgene containing Cre-responsive loxP sequence; brain-specific Cre expression | 960 (CTCGA-interrupted) |
Ribonuclear foci; MBNL1 sequestration; AS alterations; learning disability; neurotransmission dysfunction; brain atrophy and aging | Does not reproduce CTG-repeat continuity; mouse model no longer available | Identify mechanisms involved in CTG-dependent neuronal degeneration | [80] | ||||||
| TREDT960I/𝛼-MHC-rtTA | Insertion of Tet-responsive expanded DMPK exons 11–15 transgene; heart-specific rtTA expression | 960 (CTCGA-interrupted) |
Ribonuclear foci; MBNL1 sequestration; CELF1 protein upregulation; AS alterations ; arrhythmias | Does not reproduce CTG-repeat continuity | Study of alteration of ion transport and action potential in cardiomyocytes expressing toxic CUG | [81][82] | ||||||
| TREDT960I/ MDAF-rtTA |
Insertion of Tet-responsive expanded DMPK exons 11–15 transgene; skeletal-muscle-specific rtTA expression | 960 (CTCGA-interrupted) |
Ribonuclear foci; MBNL1 sequestration; CELF1 protein upregulation; AS alterations; muscle wasting and myopathy | Does not reproduce CTG-repeat continuity | Study the mechanisms of CUG-repeat-induced muscle tissue loss | [83] | ||||||
Abbreviations: AS = alternative splicing; ChP = brain choroid plexus; CMVβA = cytomegalovirus enhancer/β-actin; GFP = green fluorescent protein; KO = knockout; MDAF = expression vector carrying regulatory sequences for the rat myosin light chain 1/3 gene; MHC = myosin heavy chain; Myo = myogenin; NADH = nicotinamide adenine dinucleotide; polyA = polyadenylation; rtTA = reverse tet transactivator.
Based on the assumption that the expanded DMPK transcript is the main cause of DM1 disease, many different mouse models expressing expanded CUG transcripts either ubiquitously or in specific tissues were generated to model the disease mechanisms (Table 1B). The multisystemic impact of CUG expansions is well recapitulated in DM200, DM300 and in DMSXL transgenic mice carrying the 3′ UTR portion or the entire human DMPK gene with CTG repeats of different lengths, the phenotype being more severe in mice with larger expansion [23][68][71]. In DM300 and DMSXL mice, transgene expression resulted in the accumulation of ribonuclear foci in various tissues and in the development of muscle weakness, behavioral abnormalities, growth retardation and perinatal mortality [23][71][72][73][74]. Recently, a mouse model constitutively expressing CTG repeats within the DMPK context was generated, which exhibited particularly high CUG expression in the heart (LC15). These mice reproduced DM1-like cardiac defects [77]. Skeletal-muscle-specific, heart-specific and brain-specific DM1-like features have been reproduced in mouse models expressing the repeat expansion in the respective tissues, either constitutively (HSALR) [76] or inducibly (EpA960 and TREDT960I mice strains) [75][78][79][80][81][82][83]. The tissue-specific phenotypes are usually strong and suitable for testing therapeutic molecules. However, at the same time, they do not recapitulate the multisystemic DM1 phenotype. In the inducible models, CTG repeats interrupted with stretches of CTCGA have been inserted in a portion of the DMPK human transgene. Interrupted repeats have the advantage of being more stable than CTG repeats, but may not exactly reproduce the human DM1 disease condition. Each of these mouse models exhibits advantages and limitations mostly depending on the temporal and spatial control of the transgene expression (detailed in Table 1). Taken together, transgenic mouse models have been crucial to understanding DM1 pathogenetic mechanisms and to testing therapeutic approaches.
References
- Johnson, N.E.; Butterfield, R.J.; Mayne, K.; Newcomb, T.; Imburgia, C.; Dunn, D.; Duval, B.; Feldkamp, M.L.; Weiss, R.B. Population-Based Prevalence of Myotonic Dystrophy Type 1 Using Genetic Analysis of Statewide Blood Screening Program. Neurology 2021, 96, e1045–e1053.
- Bird, T.D. Myotonic Dystrophy Type 1. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1165/ (accessed on 21 March 2022).
- Harper, P.; van Engelen, B.; Eymard, B.; Wilcox, D. Myotonic Dystrophy. Present management, future therapy; Oxford University Press: Oxford, UK, 2004; ISBN 9780198527824.
- Meola, G.; Cardani, R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim. Et Biophys. Acta 2015, 1852, 594–606.
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 68, 799–808.
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barcelo, J.; O’Hoy, K.; et al. Myotonic dystrophy mutation: An unstable CTG repeat in the 3′ untranslated region of the gene. Science 1992, 255, 1253–1255.
- Ashizawa, T.; Dunne, C.J.; Dubel, J.R.; Perryman, M.B.; Epstein, H.F.; Boerwinkle, E.; Hejtmancik, J.F. Anticipation in myotonic dystrophy. I. Statistical verification based on clinical and haplotype findings. Neurology 1992, 42, 1871–1877.
- Overend, G.; Legare, C.; Mathieu, J.; Bouchard, L.; Gagnon, C.; Monckton, D.G. Allele length of the DMPK CTG repeat is a predictor of progressive myotonic dystrophy type 1 phenotypes. Hum. Mol. Genet. 2019, 28, 2245–2254.
- Miller, J.N.; van der Plas, E.; Hamilton, M.; Koscik, T.R.; Gutmann, L.; Cumming, S.A.; Monckton, D.G.; Nopoulos, P.C. Variant repeats within the DMPK CTG expansion protect function in myotonic dystrophy type 1. Neurol. Genet. 2020, 6, e504.
- Andre, L.M.; Ausems, C.R.M.; Wansink, D.G.; Wieringa, B. Abnormalities in Skeletal Muscle Myogenesis, Growth, and Regeneration in Myotonic Dystrophy. Front. Neurol. 2018, 9, 368.
- Visconti, V.V.; Centofanti, F.; Fittipaldi, S.; Macri, E.; Novelli, G.; Botta, A. Epigenetics of Myotonic Dystrophies: A Minireview. Int. J. Mol. Sci. 2021, 22, 12594.
- Alwazzan, M.; Newman, E.; Hamshere, M.G.; Brook, J.D. Myotonic dystrophy is associated with a reduced level of RNA from the DMWD allele adjacent to the expanded repeat. Hum. Mol. Genet. 1999, 8, 1491–1497.
- Thornton, C.A.; Wymer, J.P.; Simmons, Z.; McClain, C.; Moxley, R.T., 3rd. Expansion of the myotonic dystrophy CTG repeat reduces expression of the flanking DMAHP gene. Nat. Genet. 1997, 16, 407–409.
- Brouwer, J.R.; Huguet, A.; Nicole, A.; Munnich, A.; Gourdon, G. Transcriptionally Repressive Chromatin Remodelling and CpG Methylation in the Presence of Expanded CTG-Repeats at the DM1 Locus. J. Nucleic Acids 2013, 2013, 567435.
- Wang, E.T.; Cody, N.A.; Jog, S.; Biancolella, M.; Wang, T.T.; Treacy, D.J.; Luo, S.; Schroth, G.P.; Housman, D.E.; Reddy, S.; et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 2012, 150, 710–724.
- Nutter, C.A.; Bubenik, J.L.; Oliveira, R.; Ivankovic, F.; Sznajder, L.J.; Kidd, B.M.; Pinto, B.S.; Otero, B.A.; Carter, H.A.; Vitriol, E.A.; et al. Cell-type-specific dysregulation of RNA alternative splicing in short tandem repeat mouse knockin models of myotonic dystrophy. Genes Dev. 2019, 33, 1635–1640.
- Kalsotra, A.; Xiao, X.; Ward, A.J.; Castle, J.C.; Johnson, J.M.; Burge, C.B.; Cooper, T.A. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl. Acad. Sci. USA 2008, 105, 20333–20338.
- Imbriano, C.; Molinari, S. Alternative Splicing of Transcription Factors Genes in Muscle Physiology and Pathology. Genes 2018, 9, 107.
- Lopez-Martinez, A.; Soblechero-Martin, P.; de-la-Puente-Ovejero, L.; Nogales-Gadea, G.; Arechavala-Gomeza, V. An Overview of Alternative Splicing Defects Implicated in Myotonic Dystrophy Type I. Genes 2020, 11, 1109.
- Kuyumcu-Martinez, N.M.; Wang, G.S.; Cooper, T.A. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell. 2007, 28, 68–78.
- Castro, A.F.; Loureiro, J.R.; Bessa, J.; Silveira, I. Antisense Transcription across Nucleotide Repeat Expansions in Neurodegenerative and Neuromuscular Diseases: Progress and Mysteries. Genes 2020, 11, 1418.
- Cho, D.H.; Thienes, C.P.; Mahoney, S.E.; Analau, E.; Filippova, G.N.; Tapscott, S.J. Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol. Cell. 2005, 20, 483–489.
- Huguet, A.; Medja, F.; Nicole, A.; Vignaud, A.; Guiraud-Dogan, C.; Ferry, A.; Decostre, V.; Hogrel, J.Y.; Metzger, F.; Hoeflich, A.; et al. Molecular, physiological, and motor performance defects in DMSXL mice carrying >1000 CTG repeats from the human DM1 locus. PLoS Genet. 2012, 8, e1003043.
- Kearse, M.G.; Wilusz, J.E. Non-AUG translation: A new start for protein synthesis in eukaryotes. Genes Dev. 2017, 31, 1717–1731.
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.; et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265.
- Koehorst, E.; Nunez-Manchon, J.; Ballester-Lopez, A.; Almendrote, M.; Lucente, G.; Arbex, A.; Chojnacki, J.; Vazquez-Manrique, R.P.; Gomez-Escribano, A.P.; Pintos-Morell, G.; et al. Characterization of RAN Translation and Antisense Transcription in Primary Cell Cultures of Patients with Myotonic Dystrophy Type 1. J. Clin. Med. 2021, 10, 5520.
- Falcone, G.; Perfetti, A.; Cardinali, B.; Martelli, F. Noncoding RNAs: Emerging players in muscular dystrophies. BioMed Res. Int. 2014, 2014, 503634.
- Perbellini, R.; Greco, S.; Sarra-Ferraris, G.; Cardani, R.; Capogrossi, M.C.; Meola, G.; Martelli, F. Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromuscul. Disord. NMD 2011, 21, 81–88.
- Perfetti, A.; Greco, S.; Cardani, R.; Fossati, B.; Cuomo, G.; Valaperta, R.; Ambrogi, F.; Cortese, A.; Botta, A.; Mignarri, A.; et al. Validation of plasma microRNAs as biomarkers for myotonic dystrophy type 1. Sci. Rep. 2016, 6, 38174.
- Rau, F.; Freyermuth, F.; Fugier, C.; Villemin, J.P.; Fischer, M.C.; Jost, B.; Dembele, D.; Gourdon, G.; Nicole, A.; Duboc, D.; et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat. Struct. Mol. Biol. 2011, 18, 840–845.
- Czubak, K.; Taylor, K.; Piasecka, A.; Sobczak, K.; Kozlowska, K.; Philips, A.; Sedehizadeh, S.; Brook, J.D.; Wojciechowska, M.; Kozlowski, P. Global Increase in Circular RNA Levels in Myotonic Dystrophy. Front. Genet. 2019, 10, 649.
- Greco, S.; Cardinali, B.; Falcone, G.; Martelli, F. Circular RNAs in Muscle Function and Disease. Int. J. Mol. Sci. 2018, 19, 3454.
- Voellenkle, C.; Perfetti, A.; Carrara, M.; Fuschi, P.; Renna, L.V.; Longo, M.; Sain, S.B.; Cardani, R.; Valaperta, R.; Silvestri, G.; et al. Dysregulation of Circular RNAs in Myotonic Dystrophy Type 1. Int. J. Mol. Sci. 2019, 20, 1938.
- Ozimski, L.L.; Sabater-Arcis, M.; Bargiela, A.; Artero, R. The hallmarks of myotonic dystrophy type 1 muscle dysfunction. Biol. Rev. Camb. Philos. Soc. 2021, 96, 716–730.
- Espinosa-Espinosa, J.; Gonzalez-Barriga, A.; Lopez-Castel, A.; Artero, R. Deciphering the Complex Molecular Pathogenesis of Myotonic Dystrophy Type 1 through Omics Studies. Int. J. Mol. Sci. 2022, 23, 1441.
- Philips, A.V.; Timchenko, L.T.; Cooper, T.A. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 1998, 280, 737–741.
- Warf, M.B.; Berglund, J.A. MBNL binds similar RNA structures in the CUG repeats of myotonic dystrophy and its pre-mRNA substrate cardiac troponin T. RNA 2007, 13, 2238–2251.
- Arandel, L.; Polay Espinoza, M.; Matloka, M.; Bazinet, A.; De Dea Diniz, D.; Naouar, N.; Rau, F.; Jollet, A.; Edom-Vovard, F.; Mamchaoui, K.; et al. Immortalized human myotonic dystrophy muscle cell lines to assess therapeutic compounds. Dis. Models Mech. 2017, 10, 487–497.
- Pantic, B.; Borgia, D.; Giunco, S.; Malena, A.; Kiyono, T.; Salvatori, S.; De Rossi, A.; Giardina, E.; Sangiuolo, F.; Pegoraro, E.; et al. Reliable and versatile immortal muscle cell models from healthy and myotonic dystrophy type 1 primary human myoblasts. Exp. Cell Res. 2016, 342, 39–51.
- Provenzano, C.; Cappella, M.; Valaperta, R.; Cardani, R.; Meola, G.; Martelli, F.; Cardinali, B.; Falcone, G. CRISPR/Cas9-Mediated Deletion of CTG Expansions Recovers Normal Phenotype in Myogenic Cells Derived from Myotonic Dystrophy 1 Patients. Mol. Ther. Nucleic Acids 2017, 9, 337–348.
- Renna, L.V.; Bose, F.; Iachettini, S.; Fossati, B.; Saraceno, L.; Milani, V.; Colombo, R.; Meola, G.; Cardani, R. Receptor and post-receptor abnormalities contribute to insulin resistance in myotonic dystrophy type 1 and type 2 skeletal muscle. PLoS ONE 2017, 12, e0184987.
- Franck, S.; Couvreu De Deckersberg, E.; Bubenik, J.L.; Markouli, C.; Barbe, L.; Allemeersch, J.; Hilven, P.; Duque, G.; Swanson, M.S.; Gheldof, A.; et al. Myotonic dystrophy type 1 embryonic stem cells show decreased myogenic potential, increased CpG methylation at the DMPK locus and RNA mis-splicing. Biol. Open 2022, 11.
- Gao, Y.; Guo, X.; Santostefano, K.; Wang, Y.; Reid, T.; Zeng, D.; Terada, N.; Ashizawa, T.; Xia, G. Genome Therapy of Myotonic Dystrophy Type 1 iPS Cells for Development of Autologous Stem Cell Therapy. Mol. Ther. 2016, 24, 1378–1387.
- Ueki, J.; Nakamori, M.; Nakamura, M.; Nishikawa, M.; Yoshida, Y.; Tanaka, A.; Morizane, A.; Kamon, M.; Araki, T.; Takahashi, M.P.; et al. Myotonic dystrophy type 1 patient-derived iPSCs for the investigation of CTG repeat instability. Sci. Rep. 2017, 7, 42522.
- Martineau, L.; Racine, V.; Benichou, S.A.; Puymirat, J. Lymphoblastoids cell lines—Derived iPSC line from a 26-year-old myotonic dystrophy type 1 patient carrying (CTG)200 expansion in the DMPK gene: CHUQi001-A. Stem Cell Res. 2018, 26, 103–106.
- Spitalieri, P.; Talarico, R.V.; Caioli, S.; Murdocca, M.; Serafino, A.; Girasole, M.; Dinarelli, S.; Longo, G.; Pucci, S.; Botta, A.; et al. Modelling the pathogenesis of Myotonic Dystrophy type 1 cardiac phenotype through human iPSC-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2018, 118, 95–109.
- Fernandez-Garibay, X.; Ortega, M.A.; Cerro-Herreros, E.; Comelles, J.; Martinez, E.; Artero, R.; Fernandez-Costa, J.M.; Ramon-Azcon, J. Bioengineeredin vitro3D model of myotonic dystrophy type 1 human skeletal muscle. Biofabrication 2021, 13.
- de Haro, M.; Al-Ramahi, I.; De Gouyon, B.; Ukani, L.; Rosa, A.; Faustino, N.A.; Ashizawa, T.; Cooper, T.A.; Botas, J. MBNL1 and CUGBP1 modify expanded CUG-induced toxicity in a Drosophila model of myotonic dystrophy type 1. Hum. Mol. Genet. 2006, 15, 2138–2145.
- Houseley, J.M.; Wang, Z.; Brock, G.J.; Soloway, J.; Artero, R.; Perez-Alonso, M.; O’Dell, K.M.; Monckton, D.G. Myotonic dystrophy associated expanded CUG repeat muscleblind positive ribonuclear foci are not toxic to Drosophila. Hum. Mol. Genet. 2005, 14, 873–883.
- Picchio, L.; Plantie, E.; Renaud, Y.; Poovthumkadavil, P.; Jagla, K. Novel Drosophila model of myotonic dystrophy type 1: Phenotypic characterization and genome-wide view of altered gene expression. Hum. Mol. Genet. 2013, 22, 2795–2810.
- Reddy, S.; Smith, D.B.; Rich, M.M.; Leferovich, J.M.; Reilly, P.; Davis, B.M.; Tran, K.; Rayburn, H.; Bronson, R.; Cros, D.; et al. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat. Genet. 1996, 13, 325–335.
- Jansen, G.; Groenen, P.J.; Bachner, D.; Jap, P.H.; Coerwinkel, M.; Oerlemans, F.; van den Broek, W.; Gohlsch, B.; Pette, D.; Plomp, J.J.; et al. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat. Genet. 1996, 13, 316–324.
- Carrell, S.T.; Carrell, E.M.; Auerbach, D.; Pandey, S.K.; Bennett, C.F.; Dirksen, R.T.; Thornton, C.A. Dmpk gene deletion or antisense knockdown does not compromise cardiac or skeletal muscle function in mice. Hum. Mol. Genet. 2016, 25, 4328–4338.
- Pandey, S.K.; Wheeler, T.M.; Justice, S.L.; Kim, A.; Younis, H.S.; Gattis, D.; Jauvin, D.; Puymirat, J.; Swayze, E.E.; Freier, S.M.; et al. Identification and characterization of modified antisense oligonucleotides targeting DMPK in mice and nonhuman primates for the treatment of myotonic dystrophy type 1. J. Pharmacol. Exp. Ther. 2015, 355, 329–340.
- Charizanis, K.; Lee, K.Y.; Batra, R.; Goodwin, M.; Zhang, C.; Yuan, Y.; Shiue, L.; Cline, M.; Scotti, M.M.; Xia, G.; et al. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 2012, 75, 437–450.
- Kanadia, R.N.; Johnstone, K.A.; Mankodi, A.; Lungu, C.; Thornton, C.A.; Esson, D.; Timmers, A.M.; Hauswirth, W.W.; Swanson, M.S. A muscleblind knockout model for myotonic dystrophy. Science 2003, 302, 1978–1980.
- Matynia, A.; Ng, C.H.; Dansithong, W.; Chiang, A.; Silva, A.J.; Reddy, S. Muscleblind1, but not Dmpk or Six5, contributes to a complex phenotype of muscular and motivational deficits in mouse models of myotonic dystrophy. PLoS ONE 2010, 5, e9857.
- Poulos, M.G.; Batra, R.; Li, M.; Yuan, Y.; Zhang, C.; Darnell, R.B.; Swanson, M.S. Progressive impairment of muscle regeneration in muscleblind-like 3 isoform knockout mice. Hum. Mol. Genet. 2013, 22, 3547–3558.
- Choi, J.; Personius, K.E.; DiFranco, M.; Dansithong, W.; Yu, C.; Srivastava, S.; Dixon, D.M.; Bhatt, D.B.; Comai, L.; Vergara, J.L.; et al. Muscleblind-Like 1 and Muscleblind-Like 3 Depletion Synergistically Enhances Myotonia by Altering Clc-1 RNA Translation. EBioMedicine 2015, 2, 1034–1047.
- Lee, K.Y.; Li, M.; Manchanda, M.; Batra, R.; Charizanis, K.; Mohan, A.; Warren, S.A.; Chamberlain, C.M.; Finn, D.; Hong, H.; et al. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med. 2013, 5, 1887–1900.
- Thomas, J.D.; Sznajder, L.J.; Bardhi, O.; Aslam, F.N.; Anastasiadis, Z.P.; Scotti, M.M.; Nishino, I.; Nakamori, M.; Wang, E.T.; Swanson, M.S. Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy. Genes Dev. 2017, 31, 1122–1133.
- Ho, T.H.; Bundman, D.; Armstrong, D.L.; Cooper, T.A. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum. Mol. Genet. 2005, 14, 1539–1547.
- Koshelev, M.; Sarma, S.; Price, R.E.; Wehrens, X.H.; Cooper, T.A. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Hum. Mol. Genet. 2010, 19, 1066–1075.
- Wang, E.T.; Ward, A.J.; Cherone, J.M.; Giudice, J.; Wang, T.T.; Treacy, D.J.; Lambert, N.J.; Freese, P.; Saxena, T.; Cooper, T.A.; et al. Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res. 2015, 25, 858–871.
- Ward, A.J.; Rimer, M.; Killian, J.M.; Dowling, J.J.; Cooper, T.A. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum. Mol. Genet. 2010, 19, 3614–3622.
- Benders, A.A.; Groenen, P.J.; Oerlemans, F.T.; Veerkamp, J.H.; Wieringa, B. Myotonic dystrophy protein kinase is involved in the modulation of the Ca2+ homeostasis in skeletal muscle cells. J. Clin. Investig. 1997, 100, 1440–1447.
- Berul, C.I.; Maguire, C.T.; Aronovitz, M.J.; Greenwood, J.; Miller, C.; Gehrmann, J.; Housman, D.; Mendelsohn, M.E.; Reddy, S. DMPK dosage alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model. J. Clin. Investig. 1999, 103, R1–R7.
- Mahadevan, M.S.; Yadava, R.S.; Yu, Q.; Balijepalli, S.; Frenzel-McCardell, C.D.; Bourne, T.D.; Phillips, L.H. Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat. Genet. 2006, 38, 1066–1070.
- Yadava, R.S.; Mandal, M.; Giese, J.M.; Rigo, F.; Bennett, C.F.; Mahadevan, M.S. Modeling muscle regeneration in RNA toxicity mice. Hum. Mol. Genet. 2021, 30, 1111–1130.
- Yadava, R.S.; Yu, Q.; Mandal, M.; Rigo, F.; Bennett, C.F.; Mahadevan, M.S. Systemic therapy in an RNA toxicity mouse model with an antisense oligonucleotide therapy targeting a non-CUG sequence within the DMPK 3′UTR RNA. Hum. Mol. Genet. 2020, 29, 1440–1453.
- Seznec, H.; Agbulut, O.; Sergeant, N.; Savouret, C.; Ghestem, A.; Tabti, N.; Willer, J.C.; Ourth, L.; Duros, C.; Brisson, E.; et al. Mice transgenic for the human myotonic dystrophy region with expanded CTG repeats display muscular and brain abnormalities. Hum. Mol. Genet. 2001, 10, 2717–2726.
- Seznec, H.; Lia-Baldini, A.S.; Duros, C.; Fouquet, C.; Lacroix, C.; Hofmann-Radvanyi, H.; Junien, C.; Gourdon, G. Transgenic mice carrying large human genomic sequences with expanded CTG repeat mimic closely the DM CTG repeat intergenerational and somatic instability. Hum. Mol. Genet. 2000, 9, 1185–1194.
- Hernandez-Hernandez, O.; Guiraud-Dogan, C.; Sicot, G.; Huguet, A.; Luilier, S.; Steidl, E.; Saenger, S.; Marciniak, E.; Obriot, H.; Chevarin, C.; et al. Myotonic dystrophy CTG expansion affects synaptic vesicle proteins, neurotransmission and mouse behaviour. Brain J. Neurol. 2013, 136, 957–970.
- Sicot, G.; Servais, L.; Dinca, D.M.; Leroy, A.; Prigogine, C.; Medja, F.; Braz, S.O.; Huguet-Lachon, A.; Chhuon, C.; Nicole, A.; et al. Downregulation of the Glial GLT1 Glutamate Transporter and Purkinje Cell Dysfunction in a Mouse Model of Myotonic Dystrophy. Cell Rep. 2017, 19, 2718–2729.
- Kimura, T.; Nakamori, M.; Lueck, J.D.; Pouliquin, P.; Aoike, F.; Fujimura, H.; Dirksen, R.T.; Takahashi, M.P.; Dulhunty, A.F.; Sakoda, S. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum. Mol. Genet. 2005, 14, 2189–2200.
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000, 289, 1769–1773.
- Tylock, K.M.; Auerbach, D.S.; Tang, Z.Z.; Thornton, C.A.; Dirksen, R.T. Biophysical mechanisms for QRS- and QTc-interval prolongation in mice with cardiac expression of expanded CUG-repeat RNA. J. Gen. Physiol. 2020, 152.
- Wang, G.S.; Kearney, D.L.; De Biasi, M.; Taffet, G.; Cooper, T.A. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J. Clin. Investig. 2007, 117, 2802–2811.
- Orengo, J.P.; Chambon, P.; Metzger, D.; Mosier, D.R.; Snipes, G.J.; Cooper, T.A. Expanded CTG repeats within the DMPK 3′ UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2008, 105, 2646–2651.
- Wang, P.Y.; Lin, Y.M.; Wang, L.H.; Kuo, T.Y.; Cheng, S.J.; Wang, G.S. Reduced cytoplasmic MBNL1 is an early event in a brain-specific mouse model of myotonic dystrophy. Hum. Mol. Genet. 2017, 26, 2247–2257.
- Rao, A.N.; Campbell, H.M.; Guan, X.; Word, T.A.; Wehrens, X.H.; Xia, Z.; Cooper, T.A. Reversible cardiac disease features in an inducible CUG repeat RNA-expressing mouse model of myotonic dystrophy. JCI Insight 2021, 6.
- Valencik, M.L.; McDonald, J.A. Codon optimization markedly improves doxycycline regulated gene expression in the mouse heart. Transgenic Res. 2001, 10, 269–275.
- Morriss, G.R.; Rajapakshe, K.; Huang, S.; Coarfa, C.; Cooper, T.A. Mechanisms of skeletal muscle wasting in a mouse model for myotonic dystrophy type 1. Hum. Mol. Genet. 2018, 27, 2789–2804.
More
Information
Subjects:
Cell & Tissue Engineering
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
05 May 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No