 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Noemi Cardenas-Rodríguez | -- | 6012 | 2022-04-26 15:44:22 | | | |
| 2 | Vivi Li | Meta information modification | 6012 | 2022-04-27 04:15:11 | | |
Video Upload Options
Epilepsy is a chronic disease that affects millions of people worldwide. Antiepileptic drugs (AEDs) are used to control seizures. Even though parts of their mechanisms of action are known, there are still components that need to be studied. Therefore, the search for novel drugs, new molecular targets, and a better understanding of the mechanisms of action of existing drugs is still crucial. Levetiracetam (LEV) is an AED that has been shown to be effective in seizure control and is well-tolerable, with a novel mechanism of action through an interaction with the synaptic vesicle protein 2A (SV2A). Moreover, LEV has other molecular targets that involve calcium homeostasis, the GABAergic system, and AMPA receptors among others, that might be integrated into a single mechanism of action that could explain the antiepileptogenic, anti-inflammatory, neuroprotective, and antioxidant properties of LEV. This puts it as a possible multitarget drug with clinical applications other than for epilepsy.
1. Introduction
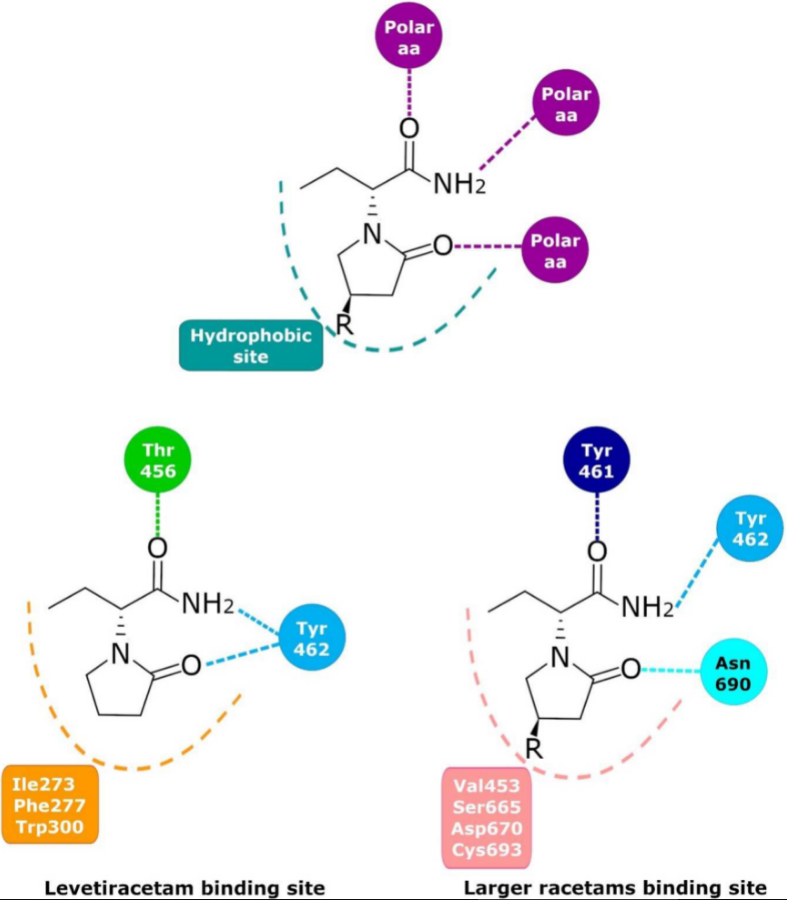
2. Levetiracetam Binding Site (LBS)
3. Molecular Mechanism
3.1. Synaptic Vesicle Protein 2A (SV2A)
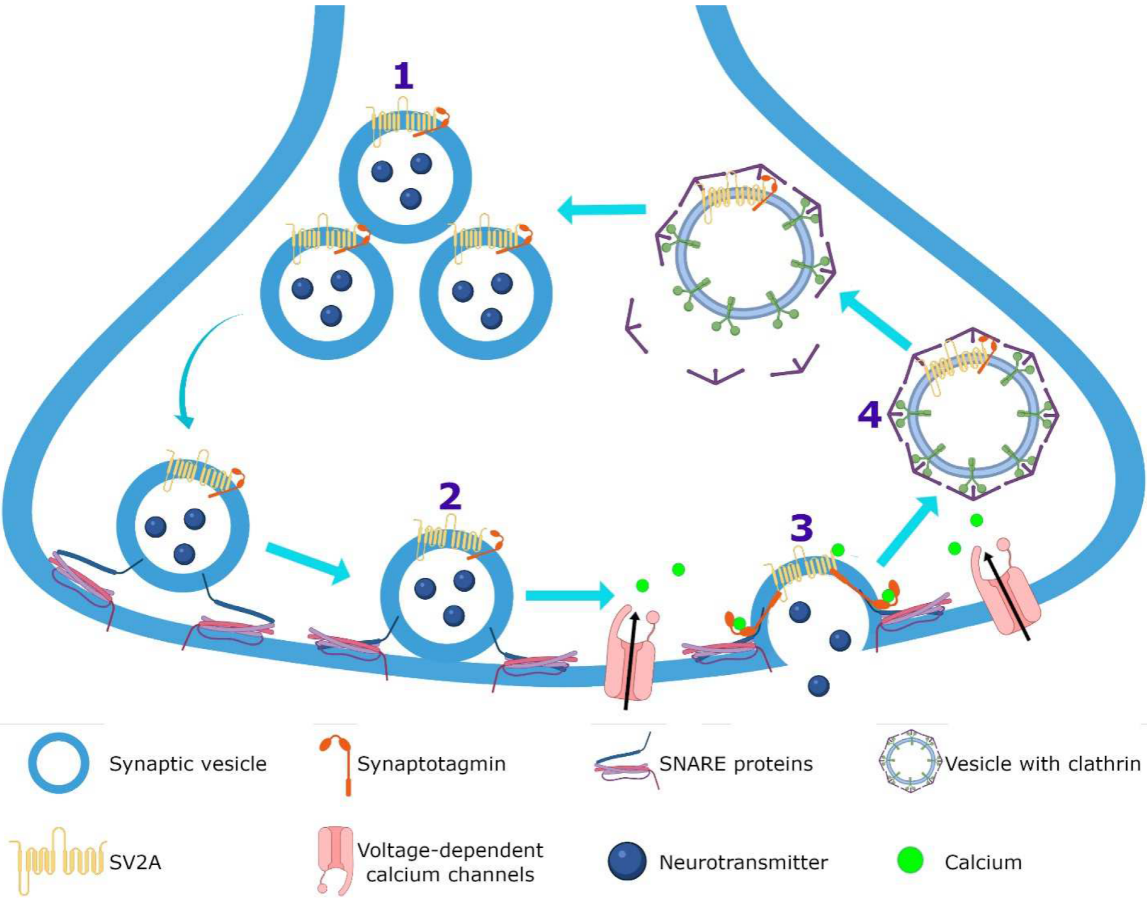
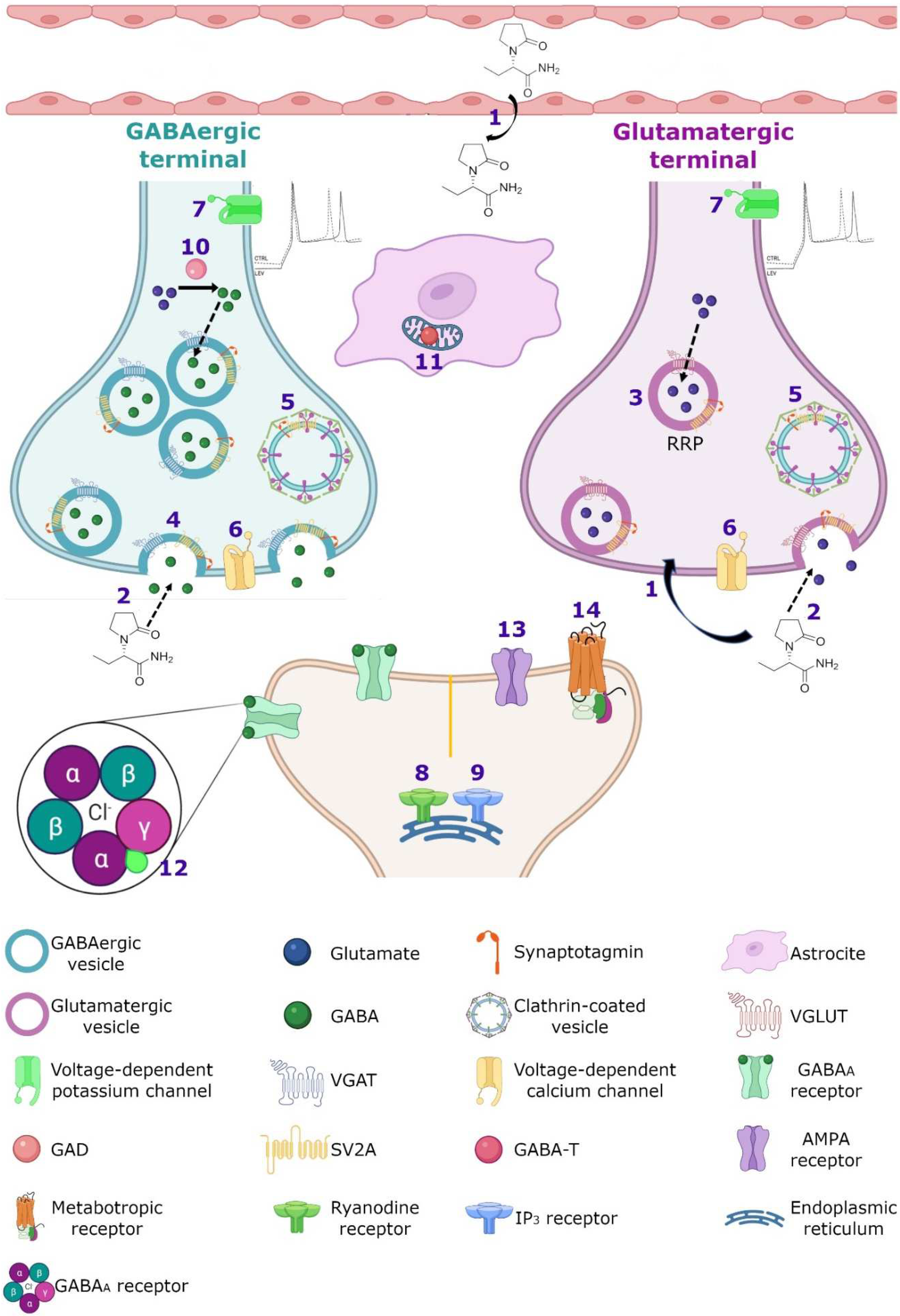
3.2. Calcium Homeostasis
3.3. GABAergic System
3.4. SV2A and GABAergic System
3.5. AMPA Receptors
3.6. Noradrenaline, Adenosine and Serotonin Receptors
3.7. Intracellular pH Regulation
3.8. Single or Integrated LEV Molecular Mechanism of Action?
4. Genetic Mechanism
Effect of Gene Polymorphisms in LEV Treatment in Clinical Studies
References
- World Health Organization (WHO). Epilepsy. Available online: https://www.who.int/news-room/fact-sheets/detail/epilepsy (accessed on 6 October 2021).
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J.J.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE official report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482.
- Fisher, R.S.; Cross, J.H.; French, J.A.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshé, S.L.; Peltola, J.; Roulet Perez, E.; et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 522–530.
- Pérez-Pérez, D.; Frías-Soria, C.L.; Rocha, L. Drug-resistant epilepsy: From multiple hypotheses to an integral explanation using preclinical resources. Epilepsy Behav. 2021, 121, 106430.
- Pitkänen, A.; Sutula, T.P. Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurol. 2002, 1, 173–181.
- Klein, P.; Friedman, A.; Hameed, M.Q.; Kaminski, R.M.; Bar-Klein, G.; Klitgaard, H.; Koepp, M.; Jozwiak, S.; Prince, D.A.; Rotenberg, A.; et al. Repurposed molecules for antiepileptogenesis: Missing an opportunity to prevent epilepsy? Epilepsia 2020, 61, 359–386.
- Alrabiah, H. Levetiracetam. Profiles Drug Subst. Excip. Relat. Methodol. 2019, 44, 167–204.
- Löscher, W.; Gillard, M.; Sands, Z.A.; Kaminski, R.M.; Klitgaard, H. Synaptic Vesicle Glycoprotein 2A Ligands in the Treatment of Epilepsy and Beyond. CNS Drugs 2016, 30, 1055–1077.
- Lynch, B.A.; Lambeng, N.; Nocka, K.; Kensel-Hammes, P.; Bajjalieh, S.M.; Matagne, A.; Fuks, B. The synaptic vesicle is the protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc. Natl. Acad. Sci. USA 2004, 101, 9861–9866.
- Crepeau, A.Z.; Treiman, D.M. Levetiracetam: A comprehensive review. Expert Rev. Neurother. 2010, 10, 159–171.
- Cortes-Altamirano, J.L.; Olmos-Hernández, A.; Bonilla-Jaime, H.; Bandala, C.; González-Maciel, A.; Alfaro-Rodríguez, A. Levetiracetam as an antiepileptic, neuroprotective, and hyperalgesic drug. Neurol. India 2016, 64, 1266–1275.
- Wong, L.C.; Freeburg, J.D.; Montouris, G.D.; Hohler, A.D. Two patients with Hashimoto’s encephalopathy and uncontrolled diabetes successfully treated with levetiracetam. J. Neurol. Sci. 2015, 348, 251–252.
- Rossi, S.; Mataluni, G.; Codecà, C.; Fiore, S.; Buttari, F.; Musella, A.; Castelli, M.; Bernardi, G.; Centonze, D. Effects of levetiracetam on chronic pain in multiple sclerosis: Results of a pilot, randomized, placebo-controlled study. Eur. J. Neurol. 2009, 16, 360–366.
- Falah, M.; Madsen, C.; Holbech, J.V.; Sindrup, S.H. A randomized, placebo-controlled trial of levetiracetam in central pain in multiple sclerosis. Eur. J. Pain 2012, 16, 860–869.
- Steinhoff, B.J.; Staack, A.M. Levetiracetam and brivaracetam: A review of evidence from clinical trials and clinical experience. Ther. Adv. Neurol. Disord. 2019, 12, 3518.
- Kenda, B.M.; Matagne, A.C.; Talaga, P.E.; Pasau, P.M.; Differding, E.; Lallemand, B.I.; Frycia, A.M.; Moureau, F.G.; Klitgaard, H.V.; Gillard, M.R.; et al. Discovery of 4-substituted pyrrolidone butanamides as new agents with significant antiepileptic activity. J. Med. Chem. 2004, 47, 530–549.
- Leclercq, K.; Matagne, A.; Provins, L.; Klitgaard, H.; Kaminski, R.M. Pharmacological Profile of the Novel Antiepileptic Drug Candidate Padsevonil: Characterization in Rodent Seizure and Epilepsy Models. J. Pharmacol. Exp. Ther. 2020, 372, 11–20.
- Noyer, M.; Gillard, M.; Matagne, A.; Hénichart, J.P.; Wülfert, E. The novel antiepileptic drug levetiracetam (ucb L059) appears to act via a specific binding site in CNS membranes. Eur. J. Pharmacol. 1995, 286, 137–146.
- Gillard, M.; Fuks, B.; Michel, P.; Vertongen, P.; Massingham, R.; Chatelain, P. Binding characteristics of ucb 30889 to levetiracetam binding sites in rat brain. Eur. J. Pharmacol. 2003, 478, 1–9.
- Rogawski, M.A. Brivaracetam: A rational drug discovery success story. Br. J. Pharmacol. 2008, 154, 1555–1557.
- Bajjalieh, S.M.; Peterson, K.; Shinghal, R.; Scheller, R.H. SV2, a brain synaptic vesicle protein homologous to bacterial transporters. Science 1992, 257, 1271–1273.
- Gillard, M.; Fuks, B.; Leclercq, K.; Matagne, A. Binding characteristics of brivaracetam, a selective, high affinity SV2A ligand in rat, mouse and human brain: Relationship to anti-convulsant properties. Eur. J. Pharmacol. 2011, 664, 36–44.
- Shi, J.; Anderson, D.; Lynch, B.A.; Castaigne, J.-G.; Foerch, P.; Lebon, F. Combining modelling and mutagenesis studies of synaptic vesicle protein 2A to identify a series of residues involved in racetam binding. Biochem. Soc. Trans. 2011, 39, 1341–1347.
- Lee, J.; Daniels, V.; Sands, Z.A.; Lebon, F.; Shi, J.; Biggin, P.C. Exploring the interaction of SV2A with racetams using homology modelling, molecular dynamics and site-directed mutagenesis. PLoS ONE 2015, 10, e0116589.
- Correa-Basurto, J.; Cuevas-Hernández, R.I.; Phillips-Farfán, B.V.; Martínez-Archundia, M.; Romo-Mancillas, A.; Ramírez-Salinas, G.L.; Pérez-González, Ó.A.; Trujillo-Ferrara, J.; Mendoza-Torreblanca, J.G. Identification of the antiepileptic racetam binding site in the synaptic vesicle protein 2A by molecular dynamics and docking simulations. Front. Cell. Neurosci. 2015, 9, 125.
- Wood, M.D.; Gillard, M. Evidence for a differential interaction of brivaracetam and levetiracetam with the synaptic vesicle 2A protein. Epilepsia 2017, 58, 255–262.
- Wood, M.D.; Sands, Z.A.; Vandenplas, C.; Gillard, M. Further evidence for a differential interaction of brivaracetam and levetiracetam with the synaptic vesicle 2A protein. Epilepsia 2018, 59, e147–e151.
- Daniels, V.; Wood, M.; Leclercq, K.; Kaminski, R.M.; Gillard, M. Modulation of the conformational state of the SV2A protein by an allosteric mechanism as evidenced by ligand binding assays. Br. J. Pharmacol. 2013, 169, 1091–1101.
- Janz, R.; Goda, Y.; Geppert, M.; Missler, M.; Südhof, T.C. SV2A and SV2B function as redundant Ca2+ regulators in neurotransmitter release. Neuron 1999, 24, 1003–1016.
- Chang, W.-P.; Südhof, T.C. SV2 renders primed synaptic vesicles competent for Ca2+ -induced exocytosis. J. Neurosci. 2009, 29, 883–897.
- Pichardo, L.A.; Contreras, I.J.; Zamudio, S.R.; Mixcoha, E.; Mendoza, J.G. Synaptic Vesicle Protein 2A as a novel pharmacological target with broad potential for new antiepileptic drugs. In Antiepileptic Drug Discovery: Novel Approaches, Methods in Pharmacology and Toxicology, 1st ed.; Talevi, A., Rocha, L., Eds.; Springer: Berlin, Germany, 2016; pp. 53–65. ISBN 13:978-1493963539.
- Crowder, K.M.; Gunther, J.M.; Jones, T.A.; Hale, B.D.; Zhang, H.Z.; Peterson, M.R.; Scheller, R.H.; Chavkin, C.; Bajjalieh, S.M. Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A). Proc. Natl. Acad. Sci. USA 1999, 96, 15268–15273.
- Custer, K.L.; Austin, N.S.; Sullivan, J.M.; Bajjalieh, S.M. Synaptic vesicle protein 2 enhances release probability at quiescent synapses. J. Neurosci. 2006, 26, 1303–1313.
- Vogl, C.; Tanifuji, S.; Danis, B.; Daniels, V.; Foerch, P.; Wolff, C.; Whalley, B.J.; Mochida, S.; Stephens, G.J. Synaptic vesicle glycoprotein 2A modulates vesicular release and calcium channel function at peripheral sympathetic synapses. Eur. J. Neurosci. 2015, 41, 398–409.
- Xu, T.; Bajjalieh, S.M. SV2 modulates the size of the readily releasable pool of secretory vesicles. Nat. Cell Biol. 2001, 3, 691–698.
- Venkatesan, K.; Alix, P.; Marquet, A.; Doupagne, M.; Niespodziany, I.; Rogister, B.; Seutin, V. Altered balance between excitatory and inhibitory inputs onto CA1 pyramidal neurons from SV2A-deficient but not SV2B-deficient mice. J. Neurosci. Res. 2012, 90, 2317–2327.
- Schivell, A.E.; Mochida, S.; Kensel-Hammes, P.; Custer, K.L.; Bajjalieh, S.M. SV2A and SV2C contain a unique synaptotagmin-binding site. Mol. Cell. Neurosci. 2005, 29, 56–64.
- Yao, J.; Nowack, A.; Kensel-Hammes, P.; Gardner, R.G.; Bajjalieh, S.M. Cotrafficking of SV2 and synaptotagmin at the synapse. J. Neurosci. 2010, 30, 5569–5578.
- Stout, K.A.; Dunn, A.R.; Hoffman, C.; Miller, G.W. The Synaptic Vesicle Glycoprotein 2: Structure, Function, and Disease Relevance. ACS Chem. Neurosci. 2019, 10, 3927–3938.
- Ciruelas, K.; Marcotulli, D.; Bajjalieh, S.M. Synaptic vesicle protein 2: A multi-faceted regulator of secretion. Semin. Cell Dev. Biol. 2019, 95, 130–141.
- Nowack, A.; Malarkey, E.B.; Yao, J.; Bleckert, A.; Hill, J.; Bajjalieh, S.M. Levetiracetam reverses synaptic deficits produced by overexpression of SV2A. PLoS ONE 2011, 6, e29560.
- Meehan, A.L.; Yang, X.; McAdams, B.D.; Yuan, L.L.; Rothman, S.M. A new mechanism for antiepileptic drug action: Vesicular entry may mediate the effects of levetiracetam. J. Neurophysiol. 2011, 106, 1227–1239.
- Mendoza-Torreblanca, J.G.; Vanoye-Carlo, A.; Phillips-Farfán, B.V.; Carmona-Aparicio, L.; Gómez-Lira, G. Synaptic vesicle protein 2A: Basic facts and role in synaptic function. Eur. J. Neurosci. 2013, 38, 3529–3539.
- Nicolas, J.-M.; Hannestad, J.; Holden, D.; Kervyn, S.; Nabulsi, N.; Tytgat, D.; Huang, Y.; Chanteux, H.; Staelens, L.; Matagne, A.; et al. Brivaracetam, a selective high-affinity synaptic vesicle protein 2A (SV2A) ligand with preclinical evidence of high brain permeability and fast onset of action. Epilepsia 2016, 57, 201–209.
- Doheny, H.C.; Ratnaraj, N.; Whittington, M.A.; Jefferys, J.G.; Patsalos, P.N. Blood and cerebrospinal fluid pharmacokinetics of the novel anticonvulsant levetiracetam (ucb L059) in the rat. Epilepsy Res. 1999, 34, 161–168.
- Patsalos, P.N. Pharmacokinetic profile of levetiracetam: Toward ideal characteristics. Pharmacol. Ther. 2000, 85, 77–85.
- Tong, X.; Patsalos, P.N. A microdialysis study of the novel antiepileptic drug levetiracetam: Extracellular pharmacokinetics and effect on taurine in rat brain. Br. J. Pharmacol. 2001, 133, 867–874.
- Kaminski, R.M.; Gillard, M.; Leclercq, K.; Hanon, E.; Lorent, G.; Dassesse, D.; Matagne, A.; Klitgaard, H. Proepileptic phenotype of SV2A-deficient mice is associated with reduced anticonvulsant efficacy of levetiracetam. Epilepsia 2009, 50, 1729–1740.
- De Groot, M.; Aronica, E.; Heimans, J.J.; Reijneveld, J.C. Synaptic vesicle protein 2A predicts response to levetiracetam in patients with glioma. Neurology 2011, 77, 532–539.
- Ohno, Y.; Ishihara, S.; Terada, R.; Kikuta, M.; Sofue, N.; Kawai, Y.; Serikawa, T.; Sasa, M. Preferential increase in the hippocampal synaptic vesicle protein 2A (SV2A) by pentylenetetrazole kindling. Biochem. Biophys. Res. Commun. 2009.
- Matveeva, E.A.; Vanaman, T.C.; Whiteheart, S.W.; Slevin, J.T. Levetiracetam prevents kindling-induced asymmetric accumulation of hippocampal 7S SNARE complexes. Epilepsia 2008, 49, 1749–1758.
- Inaba, T.; Miyamoto, N.; Hira, K.; Ueno, Y.; Yamashiro, K.; Watanabe, M.; Shimada, Y.; Hattori, N.; Urabe, T. Protective Role of Levetiracetam Against Cognitive Impairment And Brain White Matter Damage in Mouse prolonged Cerebral Hypoperfusion. Neuroscience 2019, 414, 255–264.
- Contreras-García, I.J.; Gómez-Lira, G.; Phillips-Farfán, B.V.; Pichardo-Macías, L.A.; García-Cruz, M.E.; Chávez-Pacheco, J.L.; Mendoza-Torreblanca, J.G. Synaptic Vesicle Protein 2A Expression in Glutamatergic Terminals Is Associated with the Response to Levetiracetam Treatment. Brain Sci. 2021, 11, 531.
- Marcotulli, D.; Fattorini, G.; Bragina, L.; Perugini, J.; Conti, F. Levetiracetam Affects Differentially Presynaptic Proteins in Rat Cerebral Cortex. Front. Cell. Neurosci. 2017, 11, 389.
- Niespodziany, I.; Klitgaard, H.; Margineanu, D.G. Levetiracetam inhibits the high-voltage-activated Ca(2+) current in pyramidal neurones of rat hippocampal slices. Neurosci. Lett. 2001, 306, 5–8.
- Costa, C.; Martella, G.; Picconi, B.; Prosperetti, C.; Pisani, A.; Di Filippo, M.; Pisani, F.; Bernardi, G.; Calabresi, P. Multiple mechanisms underlying the neuroprotective effects of antiepileptic drugs against in vitro ischemia. Stroke 2006, 37, 1319–1326.
- Pisani, A.; Bonsi, P.; Martella, G.; De Persis, C.; Costa, C.; Pisani, F.; Bernardi, G.; Calabresi, P. Intracellular calcium increase in epileptiform activity: Modulation by levetiracetam and lamotrigine. Epilepsia 2004, 45, 719–728.
- Lukyanetz, E.A.; Shkryl, V.M.; Kostyuk, P.G. Selective blockade of N-type calcium channels by levetiracetam. Epilepsia 2002, 43, 9–18.
- Yan, H.-D.; Ishihara, K.; Seki, T.; Hanaya, R.; Kurisu, K.; Arita, K.; Serikawa, T.; Sasa, M. Inhibitory effects of levetiracetam on the high-voltage-activated L-type Ca2+ channels in hippocampal CA3 neurons of spontaneously epileptic rat (SER). Brain Res. Bull. 2013, 90, 142–148.
- Deshpande, L.S.; Delorenzo, R.J. Mechanisms of levetiracetam in the control of status epilepticus and epilepsy. Front. Neurol. 2014, 5, 11.
- Vogl, C.; Mochida, S.; Wolff, C.; Whalley, B.J.; Stephens, G.J. The synaptic vesicle glycoprotein 2A ligand levetiracetam inhibits presynaptic Ca2+ channels through an intracellular pathway. Mol. Pharmacol. 2012, 82, 199–208.
- Angehagen, M.; Margineanu, D.G.; Ben-Menachem, E.; Rönnbäck, L.; Hansson, E.; Klitgaard, H. Levetiracetam reduces caffeine-induced Ca2+ transients and epileptiform potentials in hippocampal neurons. Neuroreport 2003, 14, 471–475.
- Nagarkatti, N.; Deshpande, L.S.; DeLorenzo, R.J. Levetiracetam inhibits both ryanodine and IP3 receptor activated calcium induced calcium release in hippocampal neurons in culture. Neurosci. Lett. 2008, 436, 289–293.
- Cataldi, M.; Lariccia, V.; Secondo, A.; di Renzo, G.; Annunziato, L. The antiepileptic drug levetiracetam decreases the inositol 1,4,5-trisphosphate-dependent I increase induced by ATP and bradykinin in PC12 cells. J. Pharmacol. Exp. Ther. 2005, 313, 720–730.
- Navidhamidi, M.; Ghasemi, M.; Mehranfard, N. Epilepsy-associated alterations in hippocampal excitability. Rev. Neurosci. 2017, 28, 307–334.
- Rigo, J.-M.; Hans, G.; Nguyen, L.; Rocher, V.; Belachew, S.; Malgrange, B.; Leprince, P.; Moonen, G.; Selak, I.; Matagne, A.; et al. The anti-epileptic drug levetiracetam reverses the inhibition by negative allosteric modulators of neuronal GABA- and glycine-gated currents. Br. J. Pharmacol. 2002, 136, 659–672.
- Doelken, M.T.; Hammen, T.; Bogner, W.; Mennecke, A.; Stadlbauer, A.; Boettcher, U.; Doerfler, A.; Stefan, H. Alterations of intracerebral γ-aminobutyric acid (GABA) levels by titration with levetiracetam in patients with focal epilepsies. Epilepsia 2010, 51, 1477–1482.
- Li, Q.; Chen, C.; Gong, T. High-field MRS study of GABA+ in patients with migraine: Response to levetiracetam treatment. Neuroreport 2018, 29, 1007–1010.
- Klitgaard, H.; Matagne, A.; Grimee, R.; Vanneste-Goemaere, J.; Margineanu, D.G. Electrophysiological, neurochemical and regional effects of levetiracetam in the rat pilocarpine model of temporal lobe epilepsy. Seizure 2003, 12, 92–100.
- Fukuyama, K.; Tanahashi, S.; Nakagawa, M.; Yamamura, S.; Motomura, E.; Shiroyama, T.; Tanii, H.; Okada, M. Levetiracetam inhibits neurotransmitter release associated with CICR. Neurosci. Lett. 2012, 518, 69–74.
- Pichardo Macías, L.A.; Ramírez Mendiola, B.A.; Contreras García, I.J.; Zamudio Hernández, S.R.; Chávez Pacheco, J.L.; Sánchez Huerta, K.B.; Mendoza Torreblanca, J.G. Effect of levetiracetam on extracellular amino acid levels in the dorsal hippocampus of rats with temporal lobe epilepsy. Epilepsy Res. 2018, 140, 111–119.
- Löscher, W.; Hönack, D.; Bloms-Funke, P. The novel antiepileptic drug levetiracetam (ucb L059) induces alterations in GABA metabolism and turnover in discrete areas of rat brain and reduces neuronal activity in substantia nigra pars reticulata. Brain Res. 1996, 735, 208–216.
- Mazzuferi, M.; Palma, E.; Martinello, K.; Maiolino, F.; Roseti, C.; Fucile, S.; Fabene, P.F.; Schio, F.; Pellitteri, M.; Sperk, G.; et al. Enhancement of GABA(A)-current run-down in the hippocampus occurs at the first spontaneous seizure in a model of temporal lobe epilepsy. Proc. Natl. Acad. Sci. USA 2010, 107, 3180–3185.
- Cifelli, P.; Palma, E.; Roseti, C.; Verlengia, G.; Simonato, M. Changes in the sensitivity of GABAA current rundown to drug treatments in a model of temporal lobe epilepsy. Front. Cell. Neurosci. 2013, 7, 108.
- Palma, E.; Ragozzino, D.; Di Angelantonio, S.; Mascia, A.; Maiolino, F.; Manfredi, M.; Cantore, G.; Esposito, V.; Di Gennaro, G.; Quarato, P.; et al. The antiepileptic drug levetiracetam stabilizes the human epileptic GABAA receptors upon repetitive activation. Epilepsia 2007, 48, 1842–1849.
- Malatynska, E.; Knapp, R.; Ikeda, M.; Yamamura, H.I. Beta-carboline interactions at the BZ-GABA receptor chloride-ionophore complex in the rat cerebral cortex. Brain Res. Bull. 1989, 22, 845–848.
- Evans, A.K.; Lowry, C.A. Pharmacology of the beta-carboline FG-7,142, a partial inverse agonist at the benzodiazepine allosteric site of the GABA A receptor: Neurochemical, neurophysiological, and behavioral effects. CNS Drug Rev. 2007, 13, 475–501.
- Kulick, C.V.; Gutherz, S.B.; Beck, V.C.; Medvedeva, N.; Soper, C.; Forcelli, P.A. Profile of anticonvulsant action of levetiracetam, tiagabine and phenobarbital against seizures evoked by DMCM (methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate) in neonatal rats. Eur. J. Pharmacol. 2014, 743, 63–68.
- Wakita, M.; Kotani, N.; Kogure, K.; Akaike, N. Inhibition of excitatory synaptic transmission in hippocampal neurons by levetiracetam involves Zn2+-dependent GABA type A receptor-mediated presynaptic modulation. J. Pharmacol. Exp. Ther. 2014, 348, 246–259.
- Buckley, K.; Kelly, R.B. Identification of a transmembrane glycoprotein specific for secretory vesicles of neural and endocrine cells. J. Cell Biol. 1985, 100, 1284–1294.
- Bajjalieh, S.M.; Frantz, G.D.; Weimann, J.M.; McConnell, S.K.; Scheller, R.H. Differential expression of synaptic vesicle protein 2 (SV2) isoforms. J. Neurosci. 1994, 14, 5223–5235.
- Bragina, L.; Fattorini, G.; Giovedí, S.; Melone, M.; Bosco, F.; Benfenati, F.; Conti, F. Analysis of Synaptotagmin, SV2, and Rab3 Expression in Cortical Glutamatergic and GABAergic Axon Terminals. Front. Cell. Neurosci. 2011, 5, 32.
- Grønborg, M.; Pavlos, N.J.; Brunk, I.; Chua, J.J.E.; Münster-Wandowski, A.; Riedel, D.; Ahnert-Hilger, G.; Urlaub, H.; Jahn, R. Quantitative comparison of glutamatergic and GABAergic synaptic vesicles unveils selectivity for few proteins including MAL2, a novel synaptic vesicle protein. J. Neurosci. 2010, 351, 981–984.
- Tokudome, K.; Okumura, T.; Terada, R.; Shimizu, S.; Kunisawa, N.; Mashimo, T.; Serikawa, T.; Sasa, M.; Ohno, Y. A Missense Mutation of the Gene Encoding Synaptic Vesicle Glycoprotein 2A (SV2A) Confers Seizure Susceptibility by Disrupting Amygdalar Synaptic GABA Release. Front. Pharmacol. 2016, 7, 210.
- Ohno, Y.; Tokudome, K. Therapeutic Role of Synaptic Vesicle Glycoprotein 2A (SV2A) in Modulating Epileptogenesis. CNS Neurol. Disord. Drug Targets 2017, 16, 463–471.
- Contreras-García, I.J.; Pichardo-Macías, L.A.; Santana-Gómez, C.E.; Sánchez-Huerta, K.; Ramírez-Hernández, R.; Gómez-González, B.; Rocha, L.; Mendoza Torreblanca, J.G. Differential expression of synaptic vesicle protein 2A after status epilepticus and during epilepsy in a lithium-pilocarpine model. Epilepsy Behav. 2018, 88, 283–294.
- Tokudome, K.; Okumura, T.; Shimizu, S.; Mashimo, T.; Takizawa, A.; Serikawa, T.; Terada, R.; Ishihara, S.; Kunisawa, N.; Sasa, M.; et al. Synaptic vesicle glycoprotein 2A (SV2A) regulates kindling epileptogenesis via GABAergic neurotransmission. Sci. Rep. 2016, 6, 27420.
- Mendoza-Torreblanca, J.G.; García-Cruz, M.E.; Sánchez-Cruz, I.; Gomez-Gonzalez, B.; Juárez-Méndez, S.; Gómez-Lira, G. Analysis of Differential Expression of Synaptic Vesicle Protein 2A in the Adult Rat Brain. Neuroscience 2019, 419, 108–120.
- Micov, A.; Tomić, M.; Popović, B.; Stepanović-Petrović, R. The antihyperalgesic effect of levetiracetam in an inflammatory model of pain in rats: Mechanism of action. Br. J. Pharmacol. 2010, 161, 384–392.
- Stepanović-Petrović, R.M.; Micov, A.M.; Tomić, M.A.; Ugrešić, N.D. The local peripheral antihyperalgesic effect of levetiracetam and its mechanism of action in an inflammatory pain model. Anesth. Analg. 2012, 115, 1457–1466.
- Carunchio, I.; Pieri, M.; Ciotti, M.T.; Albo, F.; Zona, C. Modulation of AMPA receptors in cultured cortical neurons induced by the antiepileptic drug levetiracetam. Epilepsia 2007, 48, 654–662.
- Hentschke, M.; Wiemann, M.; Hentschke, S.; Kurth, I.; Hermans-Borgmeyer, I.; Seidenbecher, T.; Jentsch, T.J.; Gal, A.; Hübner, C.A. Mice with a targeted disruption of the Cl-/HCO3- exchanger AE3 display a reduced seizure threshold. Mol. Cell. Biol. 2006, 26, 182–191.
- Svichar, N.; Esquenazi, S.; Chen, H.-Y.; Chesler, M. Preemptive regulation of intracellular pH in hippocampal neurons by a dual mechanism of depolarization-induced alkalinization. J. Neurosci. 2011, 31, 6997–7004.
- Sander, T.; Toliat, M.R.; Heils, A.; Leschik, G.; Becker, C.; Rüschendorf, F.; Rohde, K.; Mundlos, S.; Nürnberg, P. Association of the 867Asp variant of the human anion exchanger 3 gene with common subtypes of idiopathic generalized epilepsy. Epilepsy Res. 2002, 51, 249–255.
- Bonnet, U.; Bingmann, D.; Speckmann, E.-J.; Wiemann, M. Levetiracetam mediates subtle pH-shifts in adult human neocortical pyramidal cells via an inhibition of the bicarbonate-driven neuronal pH-regulation—Implications for excitability and plasticity modulation. Brain Res. 2019, 1710, 146–156.
- Leniger, T.; Thöne, J.; Bonnet, U.; Hufnagel, A.; Bingmann, D.; Wiemann, M. Levetiracetam inhibits Na+-dependent Cl-/HCO3- exchange of adult hippocampal CA3 neurons from guinea-pigs. Br. J. Pharmacol. 2004, 142, 1073–1080.
- Gu, J.; Lynch, B.A.; Anderson, D.; Klitgaard, H.; Lu, S.; Elashoff, M.; Ebert, U.; Potschka, H.; Löscher, W. The antiepileptic drug levetiracetam selectively modifies kindling-induced alterations in gene expression in the temporal lobe of rats. Eur. J. Neurosci. 2004, 19, 334–345.
- Husum, H.; Bolwig, T.G.; Sánchez, C.; Mathé, A.A.; Hansen, S.L. Levetiracetam prevents changes in levels of brain-derived neurotrophic factor and neuropeptide Y mRNA and of Y1- and Y5-like receptors in the hippocampus of rats undergoing amygdala kindling: Implications for antiepileptogenic and mood-stabilizing proper. Epilepsy Behav. 2004, 5, 204–215.
- Christensen, K.V.; Leffers, H.; Watson, W.P.; Sánchez, C.; Kallunki, P.; Egebjerg, J. Levetiracetam attenuates hippocampal expression of synaptic plasticity-related immediate early and late response genes in amygdala-kindled rats. BMC Neurosci. 2010, 11, 9.
- Kim, J.-E.; Choi, H.-C.; Song, H.-K.; Jo, S.-M.; Kim, D.-S.; Choi, S.-Y.; Kim, Y.-I.; Kang, T.-C. Levetiracetam inhibits interleukin-1 beta inflammatory responses in the hippocampus and piriform cortex of epileptic rats. Neurosci. Lett. 2010, 471, 94–99.
- Rassu, M.; Biosa, A.; Galioto, M.; Fais, M.; Sini, P.; Greggio, E.; Piccoli, G.; Crosio, C.; Iaccarino, C. Levetiracetam treatment ameliorates LRRK2 pathological mutant phenotype. J. Cell. Mol. Med. 2019, 23, 8505–8510.
- Kovacevic, J.; Maroteaux, G.; Schut, D.; Loos, M.; Dubey, M.; Pitsch, J.; Remmelink, E.; Koopmans, B.; Crowley, J.; Cornelisse, L.N.; et al. Protein instability, haploinsufficiency, and cortical hyper-excitability underlie STXBP1 encephalopathy. Brain 2018, 141, 1350–1374.
- Dilena, R.; Striano, P.; Traverso, M.; Viri, M.; Cristofori, G.; Tadini, L.; Barbieri, S.; Romeo, A.; Zara, F. Dramatic effect of levetiracetam in early-onset epileptic encephalopathy due to STXBP1 mutation. Brain Dev. 2016, 38, 128–131.
- Parveen, B.; Tripathi, M.; Vohora, D. A Cross-Sectional Study to Assess the Modulation of Wnt Inhibitors following Anti-Epileptic Drug Therapy and their Correlation with Vitamin D and Receptor Activator of Nuclear Factor κ B Ligand in Indian Women with Epilepsy. Basic Clin. Pharmacol. Toxicol. 2018, 123, 271–276.
- Lange, F.; Weßlau, K.; Porath, K.; Hörnschemeyer, J.; Bergner, C.; Krause, B.J.; Mullins, C.S.; Linnebacher, M.; Köhling, R.; Kirschstein, T. AMPA receptor antagonist perampanel affects glioblastoma cell growth and glutamate release in vitro. PLoS ONE 2019, 14, e0211644.
- Niidome, K.; Taniguchi, R.; Yamazaki, T.; Tsuji, M.; Itoh, K.; Ishihara, Y. FosL1 Is a Novel Target of Levetiracetam for Suppressing the Microglial Inflammatory Reaction. Int. J. Mol. Sci. 2021, 22, 10962.
- Hassel, B.; Taubøll, E.; Shaw, R.; Gjerstad, L.; Dingledine, R. Region-specific changes in gene expression in rat brain after chronic treatment with levetiracetam or phenytoin. Epilepsia 2010, 51, 1714–1720.
- Sills, G.J.; Rogawski, M.A. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 2020, 168, 107966.
- Belcastro, V.; Pierguidi, L.; Tambasco, N. Levetiracetam in brain ischemia: Clinical implications in neuroprotection and prevention of post-stroke epilepsy. Brain Dev. 2011, 33, 289–293.
- Rakhade, S.N.; Shah, A.K.; Agarwal, R.; Yao, B.; Asano, E.; Loeb, J.A. Activity-dependent gene expression correlates with interictal spiking in human neocortical epilepsy. Epilepsia 2007, 48 (Suppl. 5), 86–95.
- Arion, D.; Sabatini, M.; Unger, T.; Pastor, J.; Alonso-Nanclares, L.; Ballesteros-Yáñez, I.; García Sola, R.; Muñoz, A.; Mirnics, K.; DeFelipe, J. Correlation of transcriptome profile with electrical activity in temporal lobe epilepsy. Neurobiol. Dis. 2006, 22, 374–387.
- Margineanu, D.G.; Matagne, A.; Kaminski, R.M.; Klitgaard, H. Effects of chronic treatment with levetiracetam on hippocampal field responses after pilocarpine-induced status epilepticus in rats. Brain Res. Bull. 2008, 77, 282–285.
- Zhao, T.; Yu, J.; Wang, T.-T.; Feng, J.; Zhao, W.-B.; Sun, L.; Yu, L.-H.; Li, H.-J.; Sun, Y. Impact of ABCB1 Polymorphism on Levetiracetam Serum Concentrations in Epileptic Uygur Children in China. Ther. Drug Monit. 2020, 42, 886–892.
- Calame, D.G.; Herman, I.; Riviello, J.J. A de novo heterozygous rare variant in SV2A causes epilepsy and levetiracetam-induced drug-resistant status epilepticus. Epilepsy Behav. Rep. 2021, 15, 100425.
- Wolking, S.; Moreau, C.; Nies, A.T.; Schaeffeler, E.; McCormack, M.; Auce, P.; Avbersek, A.; Becker, F.; Krenn, M.; Møller, R.S.; et al. Testing association of rare genetic variants with resistance to three common antiseizure medications. Epilepsia 2020, 61, 657–666.
- Grimminger, T.; Pernhorst, K.; Surges, R.; Niehusmann, P.; Priebe, L.; von Lehe, M.; Hoffmann, P.; Cichon, S.; Schoch, S.; Becker, A.J. Levetiracetam resistance: Synaptic signatures & corresponding promoter SNPs in epileptic hippocampi. Neurobiol. Dis. 2013, 60, 115–125.
- Helmstaedter, C.; Mihov, Y.; Toliat, M.R.; Thiele, H.; Nuernberg, P.; Schoch, S.; Surges, R.; Elger, C.E.; Kunz, W.S.; Hurlemann, R. Genetic variation in dopaminergic activity is associated with the risk for psychiatric side effects of levetiracetam. Epilepsia 2013, 54, 36–44.


