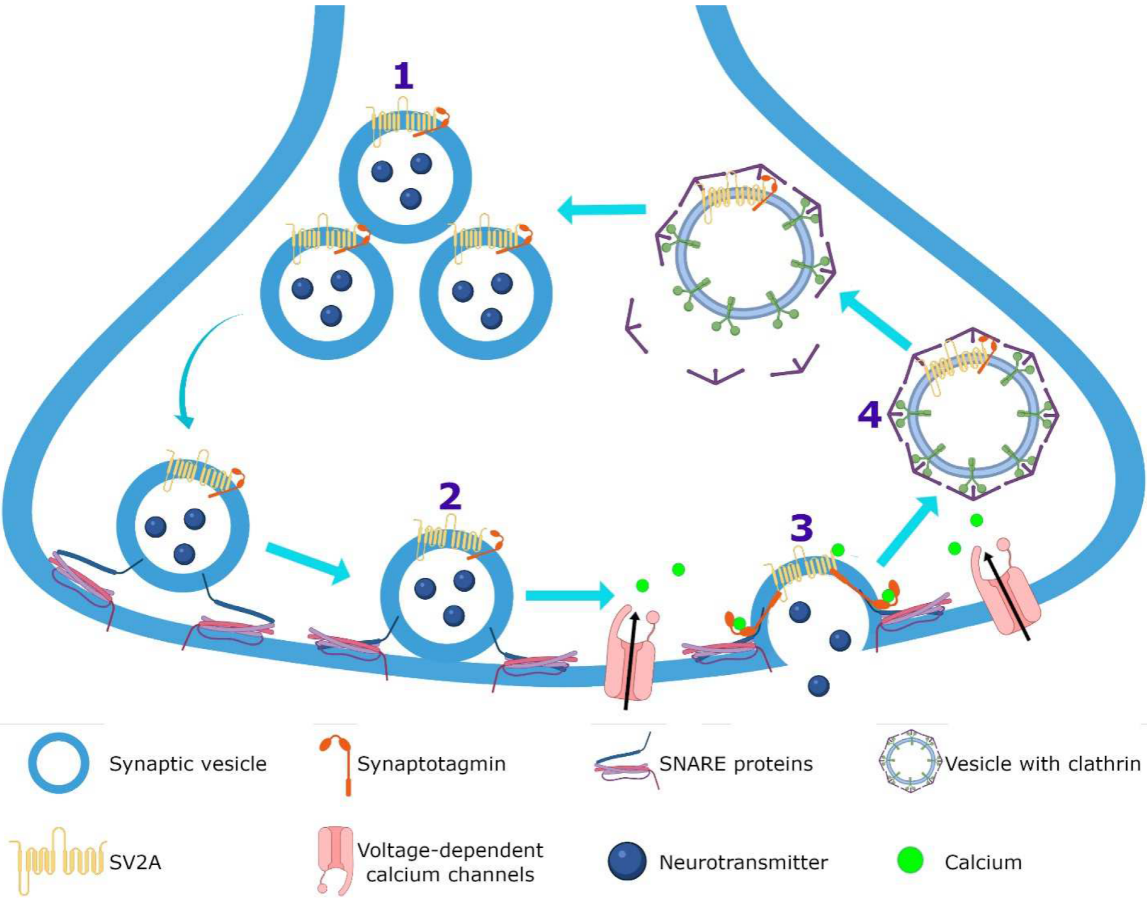
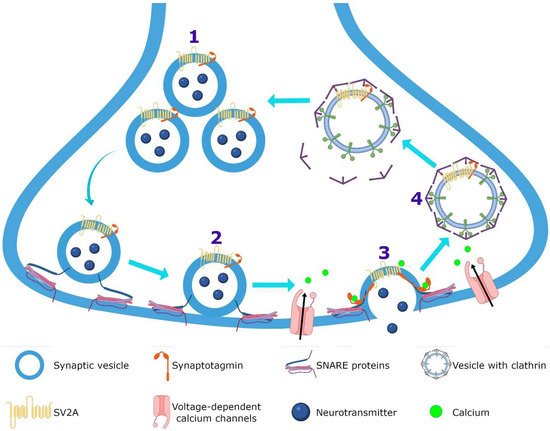
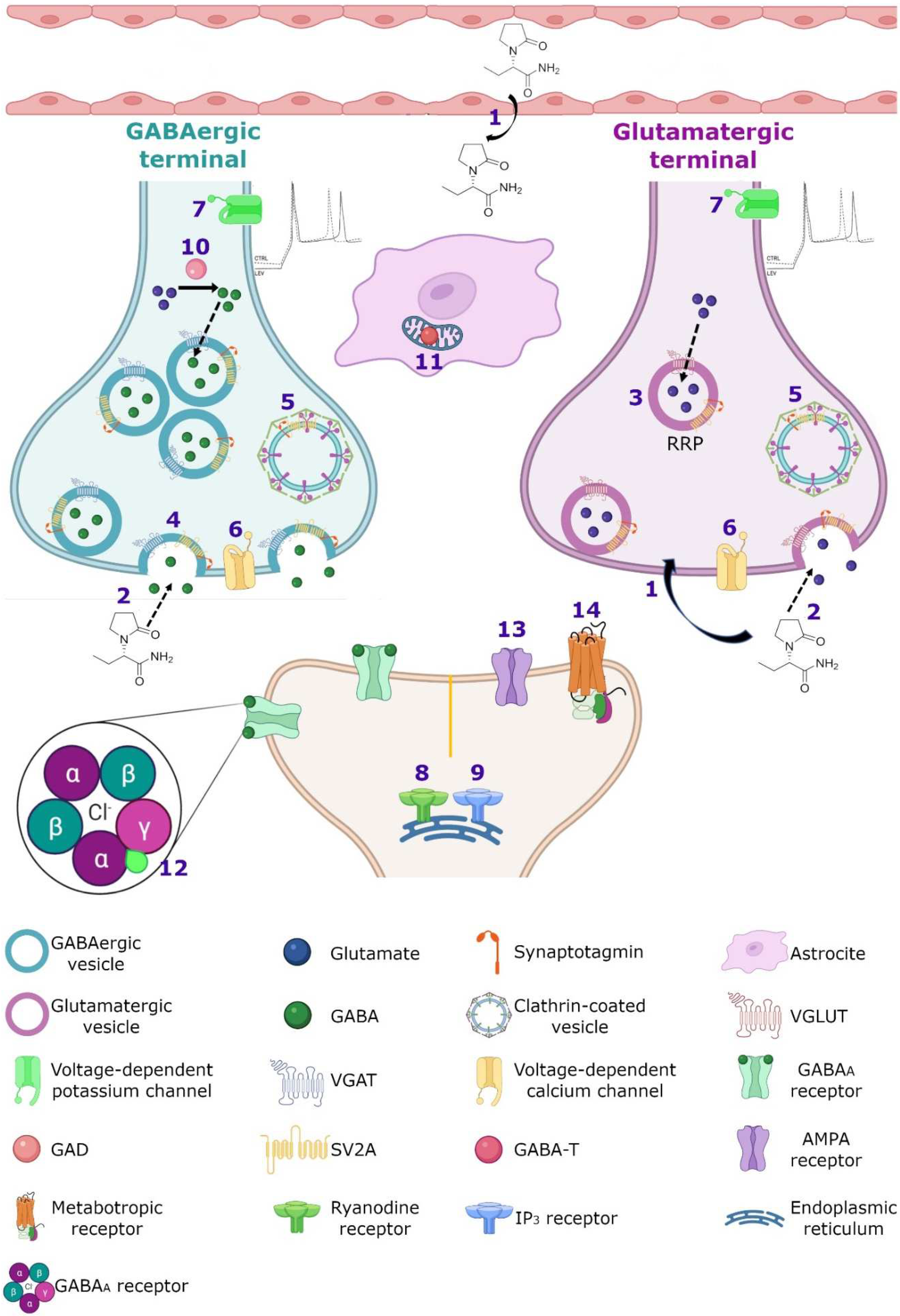
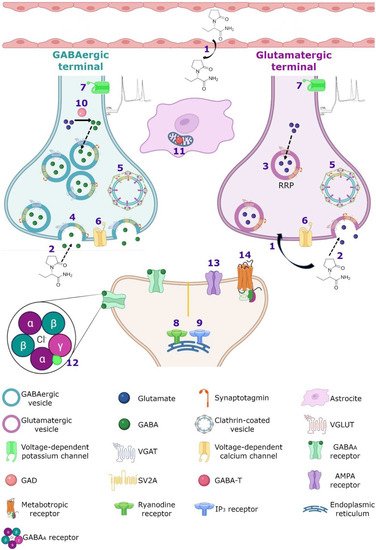
Figure 34. Hypothetical integrated molecular mechanisms of action of LEV. (
1) LEV diffuses throughout the blood–brain barrier and neuron membrane or enters during (
2) exocytosis and endocytosis processes, subsequently exerts its action by various mechanisms. (
3) LEV could decrease the function of SV2A during vesicular priming and thus diminishes the readily releasable pool and therefore the release of neurotransmitters (purple terminal). Another possibility is that LEV stabilizes SV2A and improves its function during (
4) exocytosis, and (
5) endocytosis modulating the expression and traffic of synaptotagmin protein. (
6) Moreover, it has been reported that LEV blocks the voltage-dependent calcium channels, decreasing the synaptic transmission. (
7) LEV reduces potassium currents inducing a decrease in the repetitive action potential generation. With respect to calcium intracellular systems, LEV reduces the calcium transients of (
8) ryanodine and (
9) IP
3 receptors. In the GABAergic system, LEV modulates the region-dependent (
10) glutamic acid decarboxylase (GAD) and increases (
11) GABA transaminase (GABA-T). In the post-synapse, LEV blocks the effect of the (
12) GABA
A receptor antagonists. In the glutamatergic synapse, LEV modulates (
13) AMPA receptors and decreases the excitatory current. (
14) Finally, LEV interacts with noradrenaline, adenosine and serotonin receptors in post-synapse involved in pain system.
Regarding the mechanism of the antiepileptic activity of LEV, most of the studies have focused on SV2A expression levels and/or mutations. SV2A KO mice showed severe seizures and died within 3 weeks
[32][112]. They also presented a reduced response to LEV treatment
[48][128]. Moreover, in the tumor and peritumoral tissues of glioma of patients with epilepsy, SV2A expression levels correlated with the clinical efficacy of LEV
[49][129]. Moreover, treatment with LEV blocked both the development of a seizure phenotype
[50][130] and increased the hippocampal expression of SV2A
[51][131] in kindled mice and in primary cultures of hippocampal neurons overexpressing SV2A
[41][121]. In other conditions, LEV treatment has also caused changes in protein expression. Inaba et al. found increases in the expression of SV2A and cAMP-triggered phosphorylation of cAMP response element binding protein (pCREB) in mice that suffered lesions in the white matter due to cerebral hypoperfusion. These animals preserved learning and memory capabilities in the Y-maze, spontaneous alternation, and novel object recognition tests
[52][132]. Recently
reswe
archers found that the SV2A expression in glutamatergic terminals was a key element for the response to LEV
[53][133]. In contrast, in the neocortex of non-epileptic rats, no significant differences were detected in SV2A protein levels in LEV-treated animals compared to controls
[54][134]. Certainly, both an increase and decrease in SV2A expression have been reported to be associated with the presence of seizures and it could be related with an effective response to LEV treatment.
3.2. Calcium Homeostasis
There is significant experimental evidence showing that LEV modulates targets related with cellular Ca
2+ which is a ubiquitous signal transduction molecule that plays a key role in the modulation of neuronal excitability and synaptic transmission. Specifically, LEV effects have been observed in voltage-gated channels and Ca
2+ signaling. Several studies have shown that LEV can block the high-voltage activated (HVA) Ca
2+ channels N-type, P/Q-type and L-type (
Figure 34). The administration of LEV (32 µM) in CA1 of rat hippocampal slices decreased significantly the neuronal HVA Ca
2+ currents
[55][135]. Other studies showed a selective LEV inhibition of N-type Ca
2+ channels in isolated striatal, neocortical, and CA1 pyramidal hippocampal neurons
[56][57][58][136,137,138]. In addition, LEV (100 µM) provoked a partial reduction in P/Q-type HVA Ca
2+ currents in the acutely isolated neocortical neurons
[57][137]. Moreover, LEV inhibited Ca
2+ entry by blocking the type Ca
2+ L-type channels in hippocampal CA3 neurons obtained from spontaneously epileptic rats
[59][139]. This effect was more potent than that in control neurons, suggesting that this may contribute to the antiepileptic effect of LEV
[59][60][139,140]. Moreover, it has been reported that LEV elicits effects on HVA Ca
2+ channels (presumably N-type) of superior cervical ganglion cholinergic neurons. Data showed that LEV inhibited synaptic transmission between these cells in a time-dependent manner, significantly reducing excitatory postsynaptic potential (EPSP) after a 1 h of application. Interestingly, intracellular LEV administration caused (after 4 to 5 min of exposition) rapid inhibition of the Ca
2+ current; this is consistent with a mechanism where LEV may interact directly with HVA Ca
2+ channels, causing a reduction in synaptic transmission
[34][61][19,114]. Furthermore, the application of LEV (100 μM) in acutely isolated hippocampal CA1 neurons from rats and guinea pigs, reduced the delayer rectifier K
+ currents by 26%, causing a decrease in the repetitive action potential generation and subsequent, leading to a slight prolongation of duration of the first action potential. Thus, LEV action may also be also related to its ability to hyperpolarize the membrane potential via K
+ channel activation
[11] (
Figure 34).
On the other hand, various studies have reported that LEV is an effective inhibitor of Ca
2+ release mediated by the two of the major systems of calcium-induced calcium release, ryanodine and inositol-3-phosphate (IP
3) receptors (
Figure 34). LEV significantly reduced the Ca
2+ transients induced by caffeine (a ryanodine receptor activator) in cultured rat hippocampal neurons
[62][63][141,142]. In addition, LEV inhibited the epileptiform effect induced by caffeine on the evoked field potentials and delayed caffeine-induced spontaneous bursting on rat hippocampal slices
[62][141]. Moreover, LEV inhibited Ca
2+ transients induced by bradykinin (BK; a stimulator of IP
3 receptor) in hippocampal neurons, causing a 74% diminution in calcium release mediated by the IP
3 receptor compared to the control
[63][142]. In PC12 rat pheochromocytoma cells, LEV decreased in a dose-dependent manner, the increase in Ca
2+ caused by the application of 1µM of bradykinin or 100 µM of ATP. The inhibitory effect of LEV was mainly exerted by IP
3-triggered Ca
2+ store depletion without reducing Ca
2+ storage into these deposits
[64][143]. Then, the ability of LEV to modulate ryanodine and IP
3 receptors demonstrated another important molecular effect of this agent on a major second messenger system in neurons.
3.3. GABAergic System
The GABA is the main inhibitory neurotransmitter of the central nervous system. Multiple AEDs act on its GABA
A receptor to increase inhibition and thereby controlling the aberrant neuronal activity and seizures
[65][144]. Regarding LEV, however, there are conflicting results concerning its effect on the GABAergic system
[66][145]. Patients with two distinct pathologies, focal epilepsy or migraine, were treated with LEV to determine if this drug modified brain GABA levels. By means of proton magnetic resonance spectroscopy, the GABA/creatinine ratio was evaluated before and during treatment of patients with epilepsy. The data showed an increase in GABA/creatinine in the occipital lobe in responder patients (those who showed 50–100% seizure reduction), while in non-responders the results were inconclusive
[67][146]. Meanwhile, migraine patients treated with LEV showed a decrease in headache frequency and intensity associated with a decrease in posterior cingulate cortex GABA levels
[68][147]. These results suggest that LEV may modulate differentially GABA levels and these results agreed with several animal studies. For example, the administration of LEV before injecting the convulsive agent pilocarpine in a murine model, protected against seizures modulating different neurotransmitter release; hence, LEV reversed alterations induced by focal to bilateral tonic-clonic seizures, increasing aspartate and reducing glutamine, GABA, and glycine levels in rat hippocampus
[69][148]. In addition, by K
+-evoked depolarization with microdialysis technique, LEV inhibited the release of biogenic amines, GABA, and L-glutamate in the medial prefrontal cortex of control rats
[70][149]; however, in epileptic rats treated with LEV for one week, the K
+-evoked depolarization induced a preferential increase in GABA levels without modifying other neurotransmitters in the rat dorsal hippocampus
[71][150]. In addition, the administration of LEV in the substantia nigra, a mainly GABAergic nucleus, showed that this anticonvulsive drug decreased the spontaneous firing of non-dopaminergic (maybe GABAergic) neurons, suggesting that the modulation of neuronal firing in GABAergic projections from the substantia nigra, could involve the activation or inhibition of neurotransmitter systems in other brain areas
[69][72][148,151].
In temporal lobe epilepsy (TLE) both in animal models and patients, a run-down current elicited by GABA, which is disease progression-dependent, has been reported
[73][74][152,153]; the repetitive activation of GABA
A receptors induces a decrease in GABAergic signaling (current) use-dependent in hippocampal and cortical neurons denominated as run-down, this desensitization of GABA
A receptor, could increase hyperexcitability and favor the occurrence of seizures
[73][74][152,153]. In oocytes microtransplanted with ionotropic GABA
A-receptors obtained from the resected hippocampus and temporal neocortex of patients with mesial TLE, as well as rats with pilocarpine-induced TLE, the run-down of the current evoked by GABA and the effect of LEV on this current were assessed
[74][75][153,154]. In chronic epilepsy, both rats and patients showed an increase in current GABA run-down in the hippocampus and cortex and LEV had a region-dependent effect
[74][75][153,154]; in the tissue of these rats, incubation with LEV did not affect the run-down current in the hippocampus, but it does attenuate it in the cortex
[74][153]. Meanwhile, in the tissue of patients, LEV inhibited the GABA-current run-down in the hippocampus and neocortex but was ineffective in the hippocampal subiculum
[75][154]. The authors argue that the differences could be because in the subiculum of mesial TLE patients, a switch is generated in GABA where it becomes an excitatory neurotransmitter, while in the hippocampus and neocortex GABA functions as the classic inhibitory neurotransmitter, another option to explain this data, is the differential subunit composition of GABA
A receptors and their phosphorylation
[75][154].
However, there are controversial results regarding the effect of LEV in the metabolism of GABA, since on in vitro assays LEV did not alter the activity of the GABA synthesizing enzyme, glutamic acid decarboxylase (GAD) or the GABA degrading enzyme and GABA-transaminase (GABA-T); however, on in vivo studies, a decrease in GAD in striatum and an increase in the hypothalamus at high doses of the drug was observed, as well as an increase in GABA-T activity in the cortex, striatum, thalamus and cerebellum
[72][151] (
Figure 34). Moreover, LEV modified GABA turnover by reducing it in the striatum but increasing it in the cortex and hippocampus
[72][151]. A reduction in GAD and GABA turnover in the striatum might disinhibit GABAergic striatal output pathways, augmenting the inhibition in their respective target regions and increasing the anticonvulsant effects of LEV
[72][151]. However, the authors postulate that those differential alterations on GAD and GABA-T activity between both models and in different regions might not be directly caused by LEV, instead as a consequence to pre or postsynaptic secondary effects
[72][151].
Another action of LEV in the GABAergic system involves the ability to reverse the inhibitory effects of the negative allosteric modulators β-carbolines and zinc on both GABA
A and glycine receptors, the two main ionotropic inhibitory receptor systems in the brain
[66][145]. β-carbolines congeners can act at the benzodiazepine recognition site of the GABA
A receptor complex to inhibit GABA-stimulated chloride conductance (inverse agonist effect)
[76][77][155,156] (
Figure 34). By using the whole cell patch-clamp technique, it was observed that LEV reversed the inhibitory effect of the methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate (DMCM, inverse agonist),
N-methyl-beta-carboline-3-carboxamide (FG7142, partial Inverse agonist), and butyl 9
H-pyrido[3,4-b]indole-3-carboxylate (β-CCB, inverse agonist) on GABA-elicited currents in hippocampal and cerebellar granule neurons
[66][145]. In addition, LEV completely abolished the inhibitory effects of DMCM and β-CCB on glycine currents of spinal neurons
[66][145]. The in vitro interaction of LEV with negative allosteric modulators of inhibitory receptors was confirmed in vivo in sound-susceptible mice; the administration of LEV (17 mg/kg) produced an important suppression of convulsions in these mice. The protective LEV effect was significantly diminished by the co-administration of FG 7142, from a dose of 5 mg/kg
[66][145]. In addition, in postnatal day 10 rat pups, treatment with LEV decreased the severity of DMCM-evoked seizures in a dose-dependent manner when administered in doses of 10 mg/kg and greater
[78][157].
Moreover, LEV completely reversed the inhibition by zinc of GABA and glycine evoked currents, in hippocampal and spinal cord neurons
[66][145]. By recording functional synaptic-boutons, it was observed that the activation of GABA
A receptors by muscimol (a selective GABA
A receptor agonist) induced the inhibition of evoked excitatory postsynaptic currents (eEPSCs); later, in the continued presence of muscimol, the addition of Zn
2+ increased the eEPSC amplitude (Zn
2+ had no effect by itself on the eEPSC). However, when LEV was applied in the continuous presence of muscimol and Zn
2+, there was a decrease in the eEPSC amplitude (also, LEV had no effect by itself on eEPSC, and in the presence of muscimol without Zn
2+). Then, LEV reversed the Zn
2+ induced suppression of GABA
A receptors, resulting in a decrease in glutamatergic excitatory transmission
[79][158]. These results suggest that, also, the antagonism of allosteric Zn
2+ modulation by LEV may be one of its mechanisms of action.
3.4. SV2A and GABAergic System
Despite of the fact that the SV2A protein is localized in all synaptic vesicles regardless of neurotransmitter content and its expression is similar on both glutamatergic and GABAergic terminals
[80][81][82][83][159,160,161,162], a strong relationship between SV2A and the GABAergic system has been observed. In a microdialysis study on rats with a mutation of the
SV2A gene (SV2A
L174Q), a decrease in depolarization-evoked GABA release in hippocampus and amygdala was shown, without modification of the levels of glutamate
[84][85][163,164]. In addition, in a rat model of SE induced by pilocarpine, an increase in hippocampal SV2A expression associated with GABAergic but no with glutamatergic terminals was reported
[86][165]. Moreover, recordings of cultured hippocampal pyramidal neurons (CA1 and CA3) from SV2A KO and SV2A/SV2B DKO mice showed a decreased frequency and amplitude of spontaneous inhibitory postsynaptic currents (sIPSCs) and an increase in the frequency of the spontaneous excitatory postsynaptic currents (sEPSCs) but without any change in their amplitude
[32][36][112,116]. These data agree with the observations of a great co-expression between SV2A and GABAergic neurons in the amygdala and hippocampus
[84][85][87][88][163,164,166,167]. Finally, systemic administration of LEV decreased the hyperalgesia, probably enhancing GABAergic neurotransmission by different pathways, but the local administration of LEV did not act on the GABA system
[89][90][168,169]. The close association between SV2A and the GABAergic system could be very important for the effects of LEV, but further studies must be completed in order to clarify this issue.
3.5. AMPA Receptors
Regarding the effect of LEV on glutamatergic receptors, few studies have been performed; in recordings of neuronal cortical cultures by whole cell patch-clamp, the application of kainate induced inward currents in all neurons, which were mediated primarily by the activation of AMPA receptors. The incubation with LEV in this culture, decreased kainate-induced currents by 26.5% and returned to basal after LEV washout, indicating a mild modulation of AMPA receptors
[91][170]. This was confirmed by the administration of cyclothiazide, an AMPA positive allosteric modulator in these cells, since LEV decreased the amplitude of currents induced by cyclothiazide as well as diminished the amplitude and frequency of mEPSC
[91][170].
3.6. Noradrenaline, Adenosine and Serotonin Receptors
LEV has also shown direct or indirect interaction with noradrenaline (α2A- and α2C), adenosine (A1), and serotonin (5-HT1B/1D) receptors contributing to its anti-hyperalgesic effect
[11]. In an intraplantar carrageenan-induced model of inflammatory pain, the antagonists CTAP (µ-opioid receptor antagonist), BRL-44408 (α2A-adrenoceptor antagonist), MK-912 (α2C-adrenoceptor antagonist), 1,-3-dypropyl-8-cyclopentylxanthine, DPCPX (adenosine A1 receptor antagonist), GR-127935 (5-HT1B/1D receptor antagonist) and bicuculline (GABA
A receptor antagonist) were injected (i.p. or intraplantarly) before LEV administration (systemic, 10–200 mg/kg or local, 200–1000 nmol/paw); subsequently, it was assessed if LEV had an effect by means of paw pressure test, over time (60–300 min)
[89][90][168,169]. The authors reported that LEV exerted dose and time-dependent anti-hyperalgesic activity. In contrast, the administration of all antagonists decreased this effect of LEV
[89][90][168,169]. These results suggest that LEV could be a promising drug for inflammatory pain in humans.
3.7. Intracellular pH Regulation
Variations in the normal intracellular pH influence diverse functions in neurons, glia, and interstitial space
[92][171]. Intracellular pH regulation is grouped into acid extrusion and acid loading. Acid extrusion is mainly accomplished by Na
+/H
+ exchangers, Na
+-dependent Cl
−/HCO
3− (chloride/bicarbonate) exchangers, and Na
+/HCO
3− (sodium/bicarbonate) co-transporters. Acid loading is mediated by Na
+-independent Cl
−/HCO
3− exchangers
[92][93][171,172]. Several studies have suggested that the Na
+-independent Cl
−/HCO
3− exchanger, AE3, may modulate seizure susceptibility
[92][94][171,173]. Moreover, it has been proposed that LEV mediates pH shifts and seizure-like activity via HCO
3− regulation. In human neocortical brain slices from patients with TLE pharmaco-resistant, LEV was associated with a subtle acidification, predominantly in more alkaline cells. This acidification was depended on the extracellular HCO
3− concentration
[95][20]. Then, since acidifications induced by LEV were based upon an inhibition of the Na
+/HCO
3− co-transporters, Na
+-dependent HCO
3− transporters and Na
+-dependent Cl
−/HCO
3− exchangers, LEV may decrease the intracellular pH by weakening the transmembrane HCO
3−-mediated acid extrusion
[95][96][20,174]. In addition, recordings from hippocampal slices treated with 4-aminopyridine (to increase neuronal excitability), showed that administration of LEV (10–100 µM), decreased the frequency of spontaneous action potentials and bursts of CA3 neurons. Both effects were reversible upon LEV washout and by incubating LEV plus the alkalinizing agent trimethylamine
[96][174]. Then, by inducing intracellular acidification, LEV may attenuate the excitatory neuronal activity, promoting the termination of epileptic activity and contributing to its anticonvulsive potency
[95][20].
3.8. Single or Integrated LEV Molecular Mechanism of Action?
Traditionally, the SV2A protein has been considered as the main therapeutic target of LEV. Löscher et al. proposed that although some cellular and molecular effects of LEV may contribute to its unique pharmacological profile, they have a modest magnitude
[8]. However, Cortes et al. pointed out that the mechanism of action of LEV comprises a cascade of effects that in the first instance, are exerted by binding to the SV2A protein, but its pharmacodynamics involve various molecular targets that must be integrated into a single mechanism of action by a single pathway
[11].
RWe
searchers agree with this last point of view and further propose that LEV is a unique antiepileptic agent that has multiple mechanisms of action that from an integrated point of view may explain not only its molecular effects but also their genetic, antiepileptic, antiepileptogenic, neuroprotective, antioxidant and anti-inflammatory actions (see below).
Figure 34 represents the hypothetical integrated molecular mechanisms of action of LEV.
4. Genetic Mechanism
The antiepileptic activity of LEV has been related, besides the SV2A expression, to modifications in the expression of diverse genes. In amygdala-kindled rats, this process was associated with an upregulation of hippocampal brain-derived neurotrophic factor (BDNF) and neuropeptide Y (NPY) mRNA levels. Treatment for 12 days with LEV clearly delayed the progression of kindling, showing a clear antiseizure effect and prevented the increase in BDNF and NPY mRNA
[97][98][175,176]. In addition, using real-time quantitative polymerase chain reaction temporal lobe expression of NPY gene and other epilepsy-related genes, such as, thyrotropin-releasing hormone (TRH) and glial fibrillary acidic protein (GFAP) were confirmed to be up-regulated in amygdala-kindled rats and partially normalized by LEV treatment
[97][175]. In another work using amygdala-kindled rats, LEV 1 h prior to the kindling stimulation attenuated the hippocampal overexpression of TNF-α and Cox-2, two genes related to inflammatory processes. The decrease in the expression of both genes was parallel to the antiseizure effect of the drug
[99][177]. LEV every day in 1 week also reduced the expression levels of interleukin-1β (IL-1β) and interleukin-1 receptor subtype I, and the associated reactive gliosis in the hippocampus and piriform cortex of epileptic rats
[100][178].
In relation of the effect of LEV with other genetic pathologic mechanisms, Rassu et al. showed that LEV treatment ameliorated the effect of pathological mutant phenotype of leucine-rich repeat kinase 2 (LRRK2), an enzyme that controls the vesicle trafficking
[101][179]. In th
eis study, LEV treatment significantly decreased the neurite shortening phenotype of mutant mice in primary neurons and in PC12 cells. LEV also diminishes the accumulation of dopamine receptor D2 (DRD2) into the Golgi areas due to the mutant expression in SH-SY5Y cells. These results indicated that LEV reverts LRRK2 G2019S-associated pathological effects and that LRRK2 and SV2A are involved in a common protein network controlled by LEV with the consequent modulation of traffic and dynamics in neurons
[101][179]. In another study, it was showed that LEV once daily for 5 days reduced the effect of presynaptic gene Stxbp1 mutations in
Stxbp1+/− mice, significantly reducing the number of spike-wave discharges. The de novo heterozygous mutations in
STXBP1/Munc18-1 gene were implicated in the development of early infantile epileptic encephalopathies
[102][180]. In a clinic case, LEV administered continuously from one month of age to 26 months of age had a dramatic efficacy in the treatment of encephalopathy refractory in a child with de novo heterozygous mutation (c.[922A>T]p.[Lys308(∗)]) in the STXBP1 gene
[103][181]. Moreover, it was shown that the administration of LEV (mean duration of 50.59 ± 37.93 months) in women with a diagnosis of epilepsy caused high serum levels of the Wnt antagonists sclerostin and DKK-1 in comparison with the healthy controls. Th
eis study showed that the LEV effect is implicated the modulation of the Wnt signaling pathway
[104][182]. In addition, it has been shown that LEV (18 and 180 μM) significantly decreases the gene expression of excitatory amino acid transporter 2, (EAAT2) in brain metastasis glioblastoma cells culture, indicating that LEV has a mechanism of action that decreases the recapture of glutamate from the extracellular space in brain cancer
[105][183]. In addition, in a mouse microglial BV-2 cell line culture, it was shown that lipopolysaccharide increases activator protein-1 (AP-1), FOS like 1, AP-1 transcription factor subunit (FosL1), MAF BZIP transcription factor F (MAFF) and Spi-C transcription factor (SPIC) mRNA levels and LEV attenuated AP-1 and FosL1 mRNA expression in this model. Therefore, the authors suggested that LEV can be a candidate for the treatment of neurological diseases that involve microglial activation
[106][184].
Neuronal activity influences gene expression and the drugs that modify it, through interference in neurotransmission. Therefore, LEV can affect gene expression
[42][107][108][122,185,186]. There is evidence that in HeLa cells, the main metabolite of LEV blocks histone deacetylases which catalyze the hydrolysis of acetyl groups from the lysine of some proteins, such as histone tails, inducing chromatin condensation and inhibiting gene transcription
[109][187]. In addition, it was observed in electrocorticography registers of epileptic patients an epileptiform electrical activity in the form of spikes, which has been associated with changes in gene expression
[110][111][188,189]. Thus, if LEV can inhibit hypersynchronous neuronal activity by reducing the epileptiform activity-induced population spikes in CA3
[69][148] and in dentate gyrus (DG)
[112][190], this suggests that LEV could modulate the expression of genes, as
reswe
archers mentioned above.
Effect of Gene Polymorphisms in LEV Treatment in Clinical Studies
Several investigators have studied the association of some gene polymorphisms with LEV treatment (with a target dose of 20–60 mg·kg
−1 daily for 3–4 weeks) in epileptic population. Zhao et al. studied the impact of adenosine-triphosphate (ATP)-binding cassette sub-family B member 1 (ABCB1) polymorphisms rs1128503 (C1236T), rs2032582 (G2677T) and rs1045642 (C3425T) in exon 26 on LEV serum levels and treatment efficacy in 245 Uygur Chinese children with epilepsy (drug resistant and drug-responsive). The authors showed that for genotype frequencies of ABCB1 G2677T/A, the GT genotype frequency was significantly different between drug-resistant and drug-responsive groups (OR = 0.484, 95%CI = 0.236–0.003,
p = 0.046). The other genotype frequencies did not significantly differ between both groups
[113][191]. In relation to the serum drug concentration of LEV and the serum drug concentration/body mass dose ratios (CDR), it was observed that rs2032582 and rs1045642 polymorphisms are significantly related to LEV concentration and CDR values. A higher LEV concentration and CDR values were found in GT, TT, GA, and AT genotype carriers of rs2032582 polymorphism in comparison with GG carriers (
p = 0.021). Higher LEV concentrations and CDR values were found in TT genotype carriers of rs1045642 compared with CC and CT carriers (
p = 0.002). The authors concluded that ABCB1 rs2032582 and rs1045642 polymorphisms may affect the therapeutic efficacy of LEV in epilepsy
[113][191].
In relation to SV2A mutations, surprisingly, in a pediatric epileptic patient with a rare de novo heterozygous variant in SV2A (NM_014849.5:c.1978G>A:p.Gly660Arg), seizures were found to worsen after treatment with increasing doses of LEV for approximately 40 days
[114][192]. Moreover, Wolking et al. investigated the genetic risk of rare variants for drug response to three AEDs (including LEV treatment for at least 1 year) through an analysis of the sequencing, genotyping, variant selection, and annotation of an epileptic cohort derived from the EpiPGX Consortium, to identify genetic biomarkers of epilepsy treatment response and adverse drug reactions
[115][193]. The individuals who met the inclusion criteria were 1622 patients with a diagnosis of focal epilepsy and genetic generalized epilepsy. The results of the gene set analysis showed a significant enrichment of protein truncating variants and splice-region variants in the SV2 gene group (SV2A and SV2B) associated with drug resistance. Thereafter, it was possible to conclude that a group of genes are related to drug kinetics or targeting in drug resistance to LEV
[115][193].
In another study, through the screening for mRNA signatures in 53 epileptic hippocampal tissue from pharmacoresistant TLE patients, abundant synapse-associated molecule mRNA signatures in LEV a priori non-responders were shown. In the promoter characterization was observed an accumulation of the rs9305614 G-allele in phosphatidylinositol
N-acetylglucosaminyltransferase promoter with activation of LBP-1 transcription factor of LEV a priori non-responders mesial TLE patients. The authors suggest that epigenetic factors predisposing for a priori LEV pharmacoresistance by transcriptional targets
[116][194]. Finally, in another study, the presence of rs1611115, rs4680 and rs1800497 polymorphisms with adverse psychotropic side effects of LEV in patients with chronic epilepsy was found to be related with a decrease in dopaminergic activity
[117][195].