Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Katarzyna Czarzasta | -- | 3469 | 2022-04-26 11:37:53 | | | |
| 2 | Rita Xu | -3 word(s) | 3466 | 2022-04-26 11:46:43 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Czarzasta, K.; Kruszewska, J.; Cudnoch-Jedrzejewska, A. Cardiac Muscle in the Course of Obesity. Encyclopedia. Available online: https://encyclopedia.pub/entry/22292 (accessed on 24 July 2026).
Czarzasta K, Kruszewska J, Cudnoch-Jedrzejewska A. Cardiac Muscle in the Course of Obesity. Encyclopedia. Available at: https://encyclopedia.pub/entry/22292. Accessed July 24, 2026.
Czarzasta, Katarzyna, Jagoda Kruszewska, Agnieszka Cudnoch-Jedrzejewska. "Cardiac Muscle in the Course of Obesity" Encyclopedia, https://encyclopedia.pub/entry/22292 (accessed July 24, 2026).
Czarzasta, K., Kruszewska, J., & Cudnoch-Jedrzejewska, A. (2022, April 26). Cardiac Muscle in the Course of Obesity. In Encyclopedia. https://encyclopedia.pub/entry/22292
Czarzasta, Katarzyna, et al. "Cardiac Muscle in the Course of Obesity." Encyclopedia. Web. 26 April, 2022.
Copy Citation
Obesity is a growing epidemiological problem, as two-thirds of the adult population are carrying excess weight. It is a risk factor for the development of cardiovascular diseases (hypertension, ischemic heart disease, myocardial infarct, and atrial fibrillation). It has also been shown that chronic obesity in people may be a cause for the development of heart failure with preserved ejection fraction (HFpEF), whose components include cellular hypertrophy, left ventricular diastolic dysfunction, and increased extracellular collagen deposition.
cardiac fibrosis
cardiac remodeling
collagen
extracellular matrix (ECM)
metalloproteinase (MMPs)
1. Introduction
Obesity today has reached pandemic proportions, as excess weight may even be affecting up to two-thirds of the adult population in developed countries and is one of the main causes of disability worldwide [1][2]. It is a well-established risk factor for the development of metabolic disorders such as insulin resistance, diabetes, dyslipidemia, as well as cardiovascular diseases such as atherosclerosis, ischemic heart disease, myocardial infarction, hypertension, and atrial fibrillation [3][4][5][6][7]. It has been postulated that it may contribute to the development of heart failure with preserved ejection fraction (HFpEF) [8], as well as heart failure with reduced ejection fraction—HFrEF (usually by increasing the risk of myocardial infarction) [8]. Obesity cardiomyopathy is a term describing heart failure in the course of severe obesity of long duration, in which left ventricular hypertrophy (LVH) is the most common finding [8]. Importantly, obesity is associated with several hemodynamic changes and metabolic, inflammatory, and neurohormonal alterations such as increased activity of both the renin–angiotensin–aldosterone system (RAAs) and the sympathetic nervous system (SNS), hyperleptinemia, and hypoadiponectinemia, which may all have an impact on heart remodeling and heart function [2][9].
Cardiac remodeling is defined as a change in the size, shape, or structure of one or more of the cardiac chambers [9]. It results from changes in both the phenotype of the cardiomyocytes and the extracellular matrix (ECM) [10]. Excessive synthesis of ECM components, especially collagen, by residential fibroblasts in the myocardium, is known as fibrosis [11]. This process is inextricably associated with constant ECM turnover and may be related to an imbalance between ECM degradation by enzymes known as metalloproteinases (MMPs) and their specific inhibitors (tissue inhibitors of metalloproteinases; TIMPs) [12]. It also may lead to disruption of the cardiac architecture, enhancing its stiffness and, therefore, deteriorating the systolic or diastolic function and facilitating the occurrence of arrhythmia [11].
2. Obesity as a Heterogenous Disorder
The definition of obesity is based on the body mass index (BMI) (calculated as the ratio of body mass in kilograms (kg) divided by the height in meters squared (m2)) and is diagnosed when the BMI value exceeds 30 kg/m2, whereas a person is described as overweight when their BMI value is 25–29.9 kg/m2 [13][14]. In children, obesity is diagnosed when the BMI value is above the 95th percentile adjusting for both age and gender [15][16]. According to WHO recommendations, the severity of obesity may be assessed byits division into classes, categorized by BMI: I class,30–34.9 kg/m2; II class, 35–39.9 kg/m2; and III class, over 40 kg/m2 [17]. The third class is also referred to as severe, extreme, or massive obesity [17]. The etiology of obesity is multifactorial, involving a complex interaction between genetical, hormonal, environmental, dietary, and behavioral factors; obesity develops when the caloric intake is disproportionately higher than the energy expenditure [18][19].
It must be acknowledged that obesity is a heterogenous disorder and there are a few possible phenotypes of obese subjects: metabolically healthy obese (MHO); metabolically abnormal obese (MAO); metabolically obese, normal weight (MONW), who are individuals characterized by a BMI < 25 kg/m2, but affected with complications such as hyperinsulinemia/insulin resistance, abdominal and visceral adiposity, unfavorable adipokine and lipid profile, and hypertension; and sarcopenic obese (SO) (whose body composition is made up of a high fat content and low muscle mass with an accompanying “normal” BMI) [20][21].
Obesity is reaching pandemic proportions and may trigger the development of serious metabolic and cardiovascular complications [22]. In fact, it is believed that the majority of the adverse effects of obesity on the cardiovascular system and mortality risk are not attributable to obesity itself but to the concomitant metabolic syndrome [3]. BMI is not always considered to be a good predictor of future health complications, as it is not a reliable measure to assess individual body fatness and adipose tissue distribution (visceral vs. subcutaneous adipose tissue) [23]. It must be remembered that excessive adipose tissue itself is also responsible for several hormonal and proinflammatory disturbances [24]. Due to several pathomechanisms, obesity may switch macrophage polarization towards the M1 phenotype, which is considered to be proinflammatory and profibrotic [8]. Furthermore, adipose tissue excess in obese individuals is associated with chronic subclinical inflammation and increased infiltrations of the immune cells [25]. Adipose tissue is also considered to be an active endocrine organ, as it secretes several hormones known as adipokines [26]. Recently, there has beena plethora of new emerging substances. Researchers include two of the most common hormones, leptin and adiponectin, whose defective signaling in obesity can contribute to myocardial fibrosis.
Several experimental models were developed to study obesity-related complications. The most typically encountered models for obesity induction are animals with genetic alterations, for example, null for the leptin gene (ob/ob), with a mutation in the leptin receptor gene (db/db, fa/fa), or fed with laboratory chow with a high percentage of fat content (high-fat diet—HFD) [27]. In animal models, similarly to obese people, obesity is accompanied by several metabolic disturbances (for example, hyperlipidemia, hyperinsulinemia, and glucose intolerance) [27].
3. Distinctive Characteristics of the Cardiovascular System in the Course of Obesity
Obesity may affect the cardiovascular system via hemodynamic (increased workload) and non-hemodynamic factors (increased activation of the sympathetic nervous system (SNS) and the renin–angiotensin–aldosterone system (RAAs), insulin resistance/hyperinsulinemia, leptin insensitivity/hyperleptinemia, reduced adiponectin concentration/hypoadiponectinemia, overexpression of the peroxisome-proliferator-activated receptor (PPAR), decreased natriuretic peptides, lipotoxicity, oxidative stress, and chronic inflammation/hypoxia) [5][28][29].
The heart of obese individuals are characterized by cardiomyocyte hypertrophy, infiltration of fat into the ECM (steatosis), and accumulation of triglycerides among the contractile elements [28][30]. All of those factors impact the left ventricle (LV) geometry [28][30]. Moreover, the myocardium in obese people is surrounded by excessive epicardial adipose tissue (EAT) [8]. It has been shown that EAT may secrete several cytokines such as tumor necrosis factor α (TNF-α), interleukin 6 (IL-6), activin A, connective tissue growth factor (cTGF), MMPs, and angiopoietin-like 2 (ANGPTL2) [26][31][32]. Moreover, there aredata showing that increased EAT in the course of obesity may facilitate cardiac fibrosis and promote the incidence of arrythmia [32][33][34][35].
Another adverse impact of obesity on the myocardium is associated with lipotoxicity [36]. Physiologically, in the healthy myocardium, free fatty acids (FFA) constitute an elementary energetic substrate (approximately 70%) for the synthesis of adenosine triphosphate (ATP) [37]. In obesity, there is high availability of those substrates in circulatory form, which can directly interact with the PPAR receptors. Activation of PPAR receptors leads to increased uptake of FFA from the blood to the cardiomyocyte due to the CD36/fatty-acid transport protein (FAT) transportation to the cell membrane and stimulation of expression of enzymes necessary for FFA removal by β-oxidation in the mitochondria [38]. It was also shown that this process may be facilitated by leptin throughthe short-term observation [39]; such a prolonged condition may induce myocardial lipid accumulation [40].
The presence of triglycerides may not appear to be toxic;nevertheless, a lipid overload that exceeds the oxidative or storage capacity of the tissue may provoke the induction of other alternative biochemical pathways, for example, towards the production of ceramides, which may preserve proapoptotic properties [36][41][42]. Moreover, FFAs promote diacylglycerol (DAG) synthesis, which may activate protein kinase C (PKC), which in turn inhibits insulin signaling via the phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt)-dependent pathway [36][38]. Lipotoxicity also contributes to the excessive production of reactive oxygen species (ROS) and impaired mitochondrial biogenesis, which favor both inflammation and cellular apoptosis [43].
In the next section, researchers describe more comprehensively the hemodynamic factors, which are the main causative factor for cardiomyocyte hypertrophy in obese individuals [28]. Non-hemodynamic factors and their contribution to the changes in the cellular and ECM components will be presented in the paragraph entitled ‘The effect of obesity on myocardial ECM expression, heart fibrosis, and cellular hypertrophy’ in Section 7.
3.1. Hemodynamic Changes Observed in Obesity
Obesity exerts several adaptive hemodynamic changes on the cardiac muscle (Figure 1) [44]. First, obesity is generally considered to be a hypercirculatory condition [45]. Elevated fat-free (lean) mass and the high activity of RAAs may be putative reasons [9][44][45]. In obese patients, increased cardiac output (CO) is observed, and as the heart rate (HR) remains normal or may be only slightly elevated, a major cause for this high CO is subjected to increased stroke volume (SV) [9]. Increased SV may also result in increased LV end-diastolic pressure, which was also detected in obese persons [9][44].
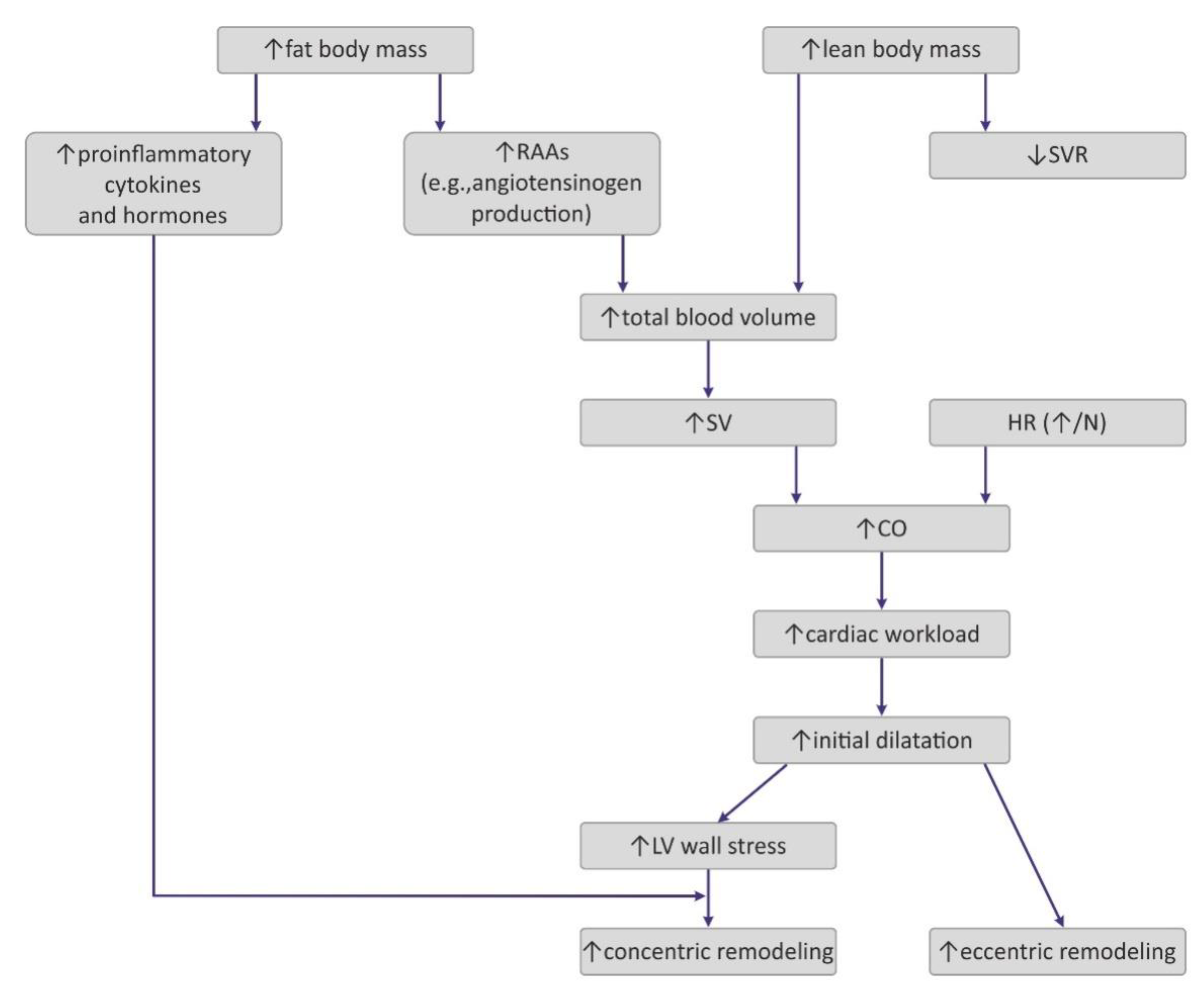
Figure 1. Pathogenesis of hemodynamic alterations in the course of obesity. RAAs—renin–angiotensin–aldosterone system; SVR—systemic vascular resistance; SV—stroke volume; HR—heart rate; CO—cardiac output; LV—left ventricle.
Interestingly, excess weight leads to decreased systemic vascular resistance (SVR), which may also lead to the augmentation of CO and LV dilatation [9]. Increased chamber volume exerts tension on the ventricular wall, according to the Law of Laplace, and such a chronic process may cause hypertrophy [29]. Such remodeling depicts the model of volume overload that contributes to eccentric hypertrophy, characterized by the lengthening of the myocytes [46].
Clinical manifestation of hemodynamic changes in the course of obesity may differ entirely depending on the distribution of the adipose tissue in the body [47]. The Dallas Heart Study carried out on 2710 participants without organic heart disease suggested that hemodynamic features such as high CO and low SVR may pertain to individuals with an excess of subcutaneous adipose tissue (SAT), especially distributed within the gluteal-femoral region, whereas increased visceral adipose tissue (VAT) was rather related to lower CO and elevated SVR [47]. Patients with excessive VAT proportions developed a concentric hypertrophy of LV (i.e., increased myocyte thickness), and also had increased LV wall thickness, increased LV mass/volume ratio, and smaller LV end-diastolic volume [46][47]. Similarly, Liu et al. showed in their study that VAT in obese patients was a better predictor of cardiac remodeling and subclinical dysfunction of LV than elevated BMI [48].
3.2. The Impact of Obesity on Myocardial Geometry and the Ejection Fraction
Left ventricular hypertrophy appears to be the most predominant observation in the hearts of obese individuals [44][49][50][51]. The results from the Framingham Heart Study, which was carried out on 3922 healthy participants, concluded that BMIwas strongly correlated with left ventricular mass, LV wall thickness, and LV internal dimensions [50]. In a different study, an increase in BMI by 1 kg/m2 and an increase in waist circumference by 1 cm increased the risk of LV hypertrophy by 5.1% and 2.6%, respectively [52]. It may be concluded that increased LV mass is commonly encountered among obese individuals and, in particular, it may be dependent on the central distribution of fat depots (abdominal fat) [53].
Nevertheless, in the literature, it is commonly discussed whether heart enlargement in the course of obesity results from concentric or rather eccentric hypertrophy [54]. A meta-analysis that included 4999 obese individuals and 6623 nonobese controls showed that eccentric hypertrophy was more frequent in obese subjects than the concentric phenotype [29][55]. This is in agreement with the initially formulated hypothesis of cardiomyopathy of obesity, based on increased cardiac output and blood volume, which in turn exerts tension on the LV wall, leading to its dilatation [56].
Today, investigators are more inclined to think that the concentric pattern may be more prevalent than initially expected [9][29] and is even independent of the occurrence of hypertension [54]. It is also believed that the distribution of fat tissue in the viscera was more correlated with concentric hypertrophy [29], as well as with higher blood pressure (BP) values in comparison with healthy, nonobese subjects, even without a diagnosis of hypertension, which may predispose to such remodeling [9]. Fat distribution and hemodynamic alterations also have an impact on cardiac remodeling, as mentioned above. Lower body SAT prompted eccentric cardiac remodeling, whereas abdominal subcutaneous adipose tissue did not have an effect on the hemodynamics. It was concluded that visceral abdominal adiposity (more often associated with the presence of metabolic syndrome) was a causative factor for concentric remodeling [47]. The existence of two different patterns of hemodynamic alterations in obese individuals may contribute to different LV remodeling (Figure 2).
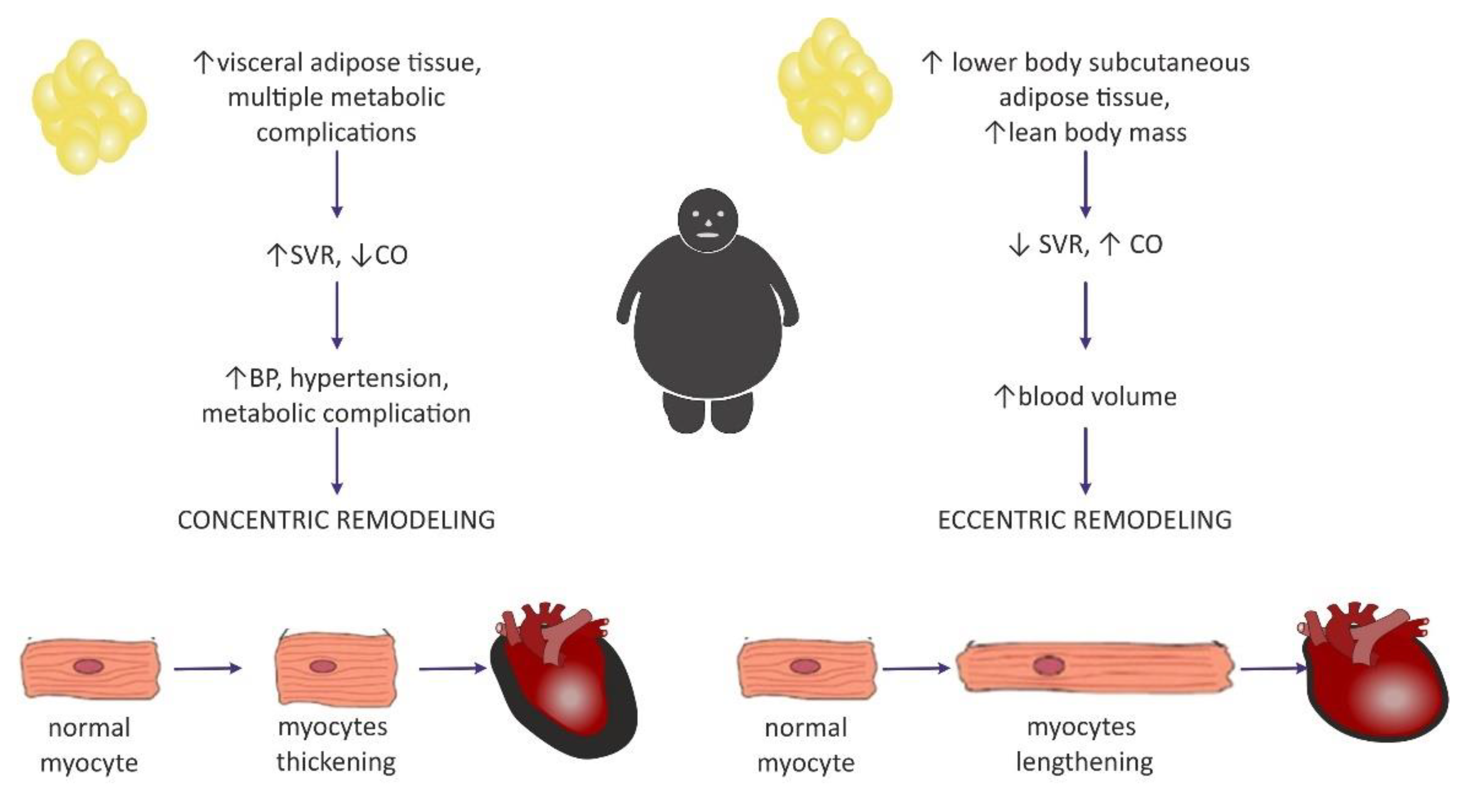
Figure 2. Two distinctive patterns of cardiac remodeling in the course of obesity and a proposition for their pathogenesis based on adipose tissue distribution. SVR—systemic vascular resistance; CO—cardiac output.
While considering LV diastolic dysfunction, it was shown that it is present in all classes of isolated obesity [1][57][58]. Moreover, its degree correlated with BMI [57]. This supports the high prevalence of HFpEF in obese individuals [59].
Data about the systolic LV function are inconsistent, as various authors have reported that the LV ejection fraction (EF) was decreased, normal, or even supernormal in obese subjects [27]. An increased EF may be observed in the early stages of obesity due to increased volume overload [57].Some authors believe that long-lasting obesity without metabolic and cardiovascular comorbidities may not be conducive to the impairment of the systolic function [1][58]. Khan et al. concluded that there is not enough evidence in clinical studies to make the claim that obese patients have left ventricular ejection fraction (LVEF) < 35% [58]. Overt systolic dysfunction may suggest the presence of concomitant heart disease, especially coronary artery disease (CAD) [1][58].
Similarly, the right ventricle (RV) in obese individuals may by characterized by a mild increase in size and wall thickness [60]. In addition, a mild dysfunction of RV was reported [8]. This may be a potential reason for the high prevalence of sleep apnea among obese individuals [9]. Nevertheless, Wong et al. concluded that increased BMI was associated with the severity of RV dysfunction in overweight and obese subjects without overt heart disease, even independent of sleep apnea [60]. Moreover, obesity may also induce the enlargement of the left atrium (LAE) as a consequence of LV diastolic dysfunction [61][62]. The large MONICA/KORA study conducted on 1212 men and women showed that the presence of obesity was the strongest predictive factor for LAE development [63] Lavie et al. presumed that obesity may constitute a risk factor for atrial fibrillation [5].
4. Myocardial Extracellular Matrix
The myocardial extracellular matrix (ECM) is a network of fibrillar collagens (mainly type I and III) and other nonfibrillar components such as glycoproteins—for example, fibronectin, proteoglycans, and glycosaminoglycans (GAG)—that surrounds the cardiac myocytes alignment and provides a connection between the cardiomyocytes, as well as between the cardiomyocytes and the surrounding vessels [64][65]. Such scaffolding converts the force generated by individual cardiomyocytes during the systole into an organized ventricular contraction and prevents cardiomyocyte slippage as well as overstretching during the diastole by providing passive stiffness [64][66]. It also influences the cardiac tissue architecture and chamber geometry [66]. Moreover, the interstitial network of connective tissues may also play a role in the mechanosensory process by intercellular signaling, such as through the collagen–integrin–cytoskeleton–myofibril connection [67]. Apart from being a cellular scaffolding, the ECM also constitutes an environment for numerous bioactive signaling molecules, such as transforming growth factor beta (TGF-β), TNF-α, angiotensin II (Ang II), and endothelin-1 (ET-1) among others [68]. They are often stored in inactive forms until they are activated in response to physiological or pathological stimuli [67].
Myocardial ECM undergoes constant turnover, approximately by 0.6% per day, physiologically [69]. Its composition is precisely regulated by MMPs and TIMPs, which are mostly synthesized by cardiac fibroblasts, as well as other cells such as cardiomyocytes, endothelial cells, and macrophages [70][71].
4.1. Collagen
Collagen is the main component of the cardiac ECM [72]. Recent morphometric evaluations of human hearts from the deceased for noncardiac reasons showed that collagen constitutes on average 15.2% of the RV, 8.6% of the interventricular septum (IVS), and 9.5% of the LV [73]. Generally, collagens, based on their structure, can be divided into two main classes: (1) fibril-forming collagens, which include the following types—I, II, III, and V; (2) nonfibrous collagens—type IV (which is the main component of the basal lamina) and type VI collagen [74]. In the cardiac ECM, fibrillar collagens types I and III are the most predominant, while collagen types IV (membrane base forming), V, and VI occur less abundantly [71]. Type I accounts for approximately 80% forms of all thick fibers and provides tensile strength in the myocardium, whereas type III collagen constitutes less than 10% of all collagens and forms a thin network of fibers that support distensibility of the heart [69][75]. The fibers are organized into the following areas: the epimysium, perimysium, and endomysium [66]. The epimysium is a sheath of connective tissue surrounding the entire muscle, whereas the perimysium surrounds groups of myocytes and the endomysium interconnects individual cells [76].
It has been shown that the amount of collagen fibers, their distribution, and organization are the determinators of heart function and alterations in its interface, both in structure and composition, may influence LV geometry and impair systolic and diastolic heart function [64][77].
Increased collagen accumulation in the ECM may appear as a sign of fibrosis within the heart muscle [71]. Beyond measurement of its protein levels, biomarkers of its synthesis and degradation are frequently assessed as indicators for collagen turnover [67][71]. Propeptides from the amino- and carboxy-terminal procollagen sides, which are cleaved in the ECM, are considered to be biomarkers of collagen synthesis. These are PICP (procollagen type I carboxy-terminal propeptide) and PINP (procollagen type I amino-terminal propeptide) for collagen type I, and their counterparts for collagen type III—PIIICP and PIIINP, and they are released in a stoichiometric manner [67][71]. During pathological ECM remodeling as well as physiological ECM turnover, collagen fibers are degraded, which is associated with the cleavage of C- and N-terminals of collagen molecules [67]. Hence, those C- and N-terminal telopeptides of collagen type I (CITP, NITP) and type III (CIIITP, NIIITP) are considered to be biomarkers of their degradation [67][71]. It is also feasible to assess the enzyme involved in collagen processing, such as prolyl-4-hydroxylase (PH4), procollagen-lysine,2-oxoglutarate 5-dioxygenase (PLOD), and lisyl oxidase (LOX) [78].
Not only does the amount of collagen have an influence on the activity of the heart muscle, but also the cross-linking of its fibers [79][80]. In most studies, the degree of cross-linking was determined by the amount of insoluble collagen versus soluble collagen in the heart [79]. The disturbances of cross-linking were observed in chronic diseases, which may be due to obesity-related comorbidities such as hypertension [81][82], chronic ventricular volume overload [83][84], diabetes [85][86] and in aging hearts [87]. Increased crosslinking may also contribute to enhanced diastolic stiffness of LV [87]. Furthermore, the reduction of cross-linking, regardless of its type and quantity may contribute to cardiac dilatation, which was observed in models of pressure-overload-induced heart failure [82].
Another factor worth considering is the ratio of type I collagen to type III collagen (I/III collagen ratio), as its increase may be responsible for left ventricle stiffness and a lower rate of relaxation [88][89]. Its elevation was observed in hypertension [79][89][90], in patients with dilated cardiomyopathy [91], obesity [92][93], and in the experimental model of myocardial infarct [94]. In diabetes, the contrary was observed, as this ratio was lower in diabetic animals and humans compared with unaffected controls [86][95].
4.2. Metalloproteinases (MMPs)
Researchers distinguish two principal types of MMPs: MMPs that are soluble in the ECM and secreted in the latent proenzyme form (proMMPs) and membrane-type metalloproteinases (MT-MMPs, such as MMP-14, also known as MT-MMP-1) that undergo processing in the cellular compartment and, subsequently, are attached to the cell membrane in the already activated form [77][96][97].
Soluble MMPs encountered in the myocardium and involved in remodeling include interstitial collagenases such as MMP-1, MMP-8, MMP-13;the stromelysins such as MMP-3; and the gelatinases such as MMP-2, MMP-9, and MMP-28 also known as epilisyn [71]. MMP-1, MMP-8, and MMP-13 degrade type I, II, and III collagens. In addition, MMP-1 degrades the basement membrane proteins [71]. The classically known gelatins, MMP-2 and MMP-9, also process many collagens, including type I, IV, and V collagen; MMP-2 additionally cleaves type III collagen [12][98]. MT1-MMP can cleave many ECM proteins, including fibronectin, laminin-1, and type I collagen [71].
Expression and activity of MMPs is tightly controlled on many different levels [97][99]. First, it is regulated on a transcriptional level by a variety of growth factors, cytokines, chemokines, hormones, cellular transformation, and interaction with extracellular matrix components. Second, most MMPs (except the membrane type) are synthesized as inactive zymogens, called proMMPs [12], which require proteolytic activation by other already active MMPs or endogenic proteases such as plasmin, urokinase-type plasminogen activator (uPA), tissue plasminogen activator (tPA), or thrombin [64]. Third, active MMPs may be inhibited directly by their most specific inhibitors, such as tissue inhibitors of metalloproteinase (TIMPs) [12].
4.3. Tissue Inhibitors of Metalloproteinase (TIMPs)
TIMPs are low-molecular-weight proteins (21–30 kDA) that create noncovalent high-affinity complexes with active MMPs in the stoichiometric 1:1 ratio [12]. To date, there have been four TIMPs (TIMP-1, -2, -3, -4) reported in the literature [100]. All four of them are expressed in the normal human heart, but their profile varies under pathological conditions [101]. Beyond their apparent inhibitory properties towards MMPs, TIMPs are also involved in several other processes and may promote cellular growth, proliferation, and apoptosis [102]. For example, it has been shown that upregulated TIMP-1 may induce collagen synthesis and its elevated serum concentration may correlate with cardiac fibrosis [103].
References
- Mahajan, R.; Lau, D.H.; Sanders, P. Impact of Obesity on Cardiac Metabolism, Fibrosis, and Function. Trends Cardiovasc. Med. 2015, 25, 119–126.
- Yumuk, V.; Frühbeck, G.; Oppert, J.M.; Woodward, E.; Toplak, H. An EASO Position Statement on Multidisciplinary Obesity Management in Adults. Obes. Facts 2014, 7, 96–101.
- Kachur, S.; Lavie, C.J.; de Schutter, A.; Milani, R.V.; Ventura, H.O. Obesity and Cardiovascular Diseases. Minerva Med. 2017, 108, 212–228.
- Karam, B.S.; Chavez-Moreno, A.; Koh, W.; Akar, J.G.; Akar, F.G. Oxidative Stress and Inflammation as Central Mediators of Atrial Fibrillation in Obesity and Diabetes. Cardiovasc. Diabetol. 2017, 16, 120.
- Lavie, C.J.; Pandey, A.; Lau, D.H.; Alpert, M.A.; Sanders, P. Obesity and Atrial Fibrillation Prevalence, Pathogenesis, and Prognosis: Effects of Weight Loss and Exercise. J. Am. Coll. Cardiol. 2017, 70, 2022–2035.
- Mendoza, M.F.; Kachur, S.M.; Lavie, C.J. Hypertension in Obesity. Curr. Opin. Cardiol. 2020, 35, 389–396.
- Obokata, M.; Reddy, Y.N.V.; Pislaru, S.V.; Melenovsky, V.; Borlaug, B.A. Evidence Supporting the Existence of a Distinct Obese Phenotype of Heart Failure with Preserved Ejection Fraction. Circulation 2017, 136, 6–19.
- Mouton, A.J.; Li, X.; Hall, M.E.; Hall, J.E. Obesity, Hypertension, and Cardiac Dysfunction Novel Roles of Immunometabolism in Macrophage Activation and Inflammation. Circ. Res. 2020, 126, 789–806.
- Alpert, M.A.; Karthikeyan, K.; Abdullah, O.; Ghadban, R. Obesity and Cardiac Remodeling in Adults: Mechanisms and Clinical Implications. Prog. Cardiovasc. Dis. 2018, 61, 114–123.
- Kehat, I.; Molkentin, J.D. Molecular Pathways Underlying Cardiac Remodeling during Pathophysiological Stimulation. Circulation 2010, 122, 2727–2735.
- Li, L.; Zhao, Q.; Kong, W. Extracellular Matrix Remodeling and Cardiac Fibrosis. Matrix Biol. 2018, 68–69, 490–506.
- Moore, L.; Fan, D.; Basu, R.; Kandalam, V.; Kassiri, Z. Tissue Inhibitor of Metalloproteinases (TIMPs) in Heart Failure. Heart Fail. Rev. 2012, 17, 693–706.
- Jensen, M.D.; Ryan, D.H.; Apovian, C.M.; Ard, J.D.; Comuzzie, A.G.; Donato, K.A.; Hu, F.B.; Hubbard, V.S.; Jakicic, J.M.; Kushner, R.F.; et al. 2013 AHA/ACC/TOS Guideline for the Management of Overweight and Obesity in Adults. Circulation 2014, 129, S102–S138.
- Bessesen, D.H. Update on Obesity. J. Clin. Endocrinol. Metab. 2008, 93, 2027–2034.
- Weir, C.B.; Jan, A. BMI Classification Percentile and Cut Off Points; StatPearls Publishing: Treasure Island, FL, USA, 2019.
- Schwartz, M.W.; Seeley, R.J.; Zeltser, L.M.; Drewnowski, A.; Ravussin, E.; Redman, L.M.; Leibel, R.L. Obesity Pathogenesis: An Endocrine Society Scientific Statement. Endocr. Rev. 2017, 38, 267–296.
- Jan, A.; Weir, C.B. BMI Classification Percentile and Cut Off Points; StatPearls: Treasure Island, FL, USA, 2021.
- Kaila, B.; Raman, M. Obesity: A Review of Pathogenesis and Management Strategies. Can. J. Gastroenterol. 2008, 22, 61–68.
- Pi-Sunyer, F.X. The Obesity Epidemic: Pathophysiology and Consequences of Obesity. Obes. Res. 2002, 10, 97S–104S.
- Mayoral, L.P.C.; Andrade, G.M.; Mayoral, E.P.C.; Huerta, T.H.; Canseco, S.P.; Rodal Canales, F.J.; Cabrera-Fuentes, H.A.; Cruz, M.M.; Pérez Santiago, A.D.; Alpuche, J.J.; et al. Obesity Subtypes, Related Biomarkers & Heterogeneity. Indian J. Med. Res. 2020, 151, 11–21.
- Lee, D.C.; Shook, R.P.; Drenowatz, C.; Blair, S.N. Physical Activity and Sarcopenic Obesity: Definition, Assessment, Prevalence and Mechanism. Future Sci. OA 2016, 2, FSO127.
- Sellayah, D.; Cagampang, F.R.; Cox, R.D. On the Evolutionary Origins of Obesity: A New Hypothesis. Endocrinology 2014, 155, 1573–1588.
- Neeland, I.J.; Poirier, P.; Després, J.P. Cardiovascular and Metabolic Heterogeneity of Obesity: Clinical Challenges and Implications for Management. Circulation 2018, 137, 1391–1406.
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358.
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2020, 10, 1607.
- Anthony, S.R.; Guarnieri, A.R.; Gozdiff, A.; Helsley, R.N.; Owens, A.P.; Tranter, M. Mechanisms Linking Adipose Tissue Inflammation to Cardiac Hypertrophy and Fibrosis. Clin. Sci. 2019, 133, 2329–2344.
- Abel, E.D.; Litwin, S.E.; Sweeney, G. Cardiac Remodeling in Obesity. Physiol. Rev. 2008, 88, 389–419.
- de Simone, G.; Izzo, R.; de Luca, N.; Gerdts, E. Left Ventricular Geometry in Obesity: Is It What We Expect? Nutr. Metab. Cardiovasc. Dis. 2013, 23, 905–912.
- Tadic, M.; Cuspidi, C. Obesity and Heart Failure with Preserved Ejection Fraction: A Paradox or Something Else? Heart Fail. Rev. 2019, 24, 379–385.
- Murdolo, G.; Angeli, F.; Reboldi, G.; di Giacomo, L.; Aita, A.; Bartolini, C.; Vedecchia, P. Left Ventricular Hypertrophy and Obesity: Only a Matter of Fat? High Blood Press. Cardiovasc. Prev. 2015, 22, 29–41.
- Hatem, S.N.; Sanders, P. Epicardial Adipose Tissue and Atrial Fibrillation. Cardiovasc. Res. 2014, 102, 205–213.
- Krishnan, A.; Chilton, E.; Raman, J.; Saxena, P.; McFarlane, C.; Trollope, A.F.; Kinobe, R.; Chilton, L. Are Interactions between Epicardial Adipose Tissue, Cardiac Fibroblasts and Cardiac Myocytes Instrumental in Atrial Fibrosis and Atrial Fibrillation? Cells 2021, 10, 2501.
- Patel, K.H.K.; Hwang, T.; Se Liebers, C.; Ng, F.S. Epicardial Adipose Tissue as a Mediator of Cardiac Arrhythmias. Am. J. Physiol.-Heart Circ. Physiol. 2022, 322, H129–H144.
- Venteclef, N.; Guglielmi, V.; Balse, E.; Gaborit, B.; Cotillard, A.; Atassi, F.; Amour, J.; Leprince, P.; Dutour, A.; Clément, K.; et al. Human Epicardial Adipose Tissue Induces Fibrosis of the Atrial Myocardium through the Secretion of Adipo-Fibrokines. Eur. Heart J. 2015, 36, 795–805.
- Abe, I.; Teshima, Y.; Kondo, H.; Kaku, H.; Kira, S.; Ikebe, Y.; Saito, S.; Fukui, A.; Shinohara, T.; Yufu, K.; et al. Association of Fibrotic Remodeling and Cytokines/Chemokines Content in Epicardial Adipose Tissue with Atrial Myocardial Fibrosis in Patients with Atrial Fibrillation. Heart Rhythm 2018, 15, 1717–1727.
- Wende, A.R.; Symons, J.D.; Abel, E.D. Mechanisms of Lipotoxicity in the Cardiovascular System. Curr. Hypertens. Rep. 2012, 14, 517–531.
- Gibb, A.A.; Hill, B.G. Metabolic Coordination of Physiological and Pathological Cardiac Remodeling. Circ. Res. 2018, 123, 107–128.
- Dirkx, E.; Schwenk, R.W.; Glatz, J.F.C.; Luiken, J.J.F.P.; van Eys, G.J.J.M. High Fat Diet Induced Diabetic Cardiomyopathy. Prostaglandins Leukot. Essent. Fat. Acids 2011, 85, 219–225.
- Karbowska, J.; Kochan, Z. Leptin as a Mediator between Obesity and Cardiac Dysfunction. Postępy Hig. I Med. Doświadczalnej 2012, 66, 267–274.
- Sletten, A.C.; Peterson, L.R.; Schaffer, J.E. Manifestations and Mechanisms of Myocardial Lipotoxicity in Obesity. J. Intern. Med. 2018, 284, 478–491.
- Zhou, Y.T.; Grayburn, P.; Karim, A.; Shimabukuro, M.; Higa, M.; Baetens, D.; Orci, L.; Unger, R.H. Lipotoxic Heart Disease in Obese Rats: Implications for Human Obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 1784–1789.
- Bielawska, A.E.; Shapiro, J.P.; Jiang, L.; Melkonyan, H.S.; Piot, C.; Wolfe, C.L.; Tomei, L.D.; Hannun, Y.A.; Umansky, S.R. Ceramide Is Involved in Triggering of Cardiomyocyte Apoptosis Induced by Ischemia and Reperfusion. Am. J. Pathol. 1997, 151, 1257–1263.
- Csonka, C.; Sárközy, M.; Pipicz, M.; Dux, L.; Csont, T. Modulation of Hypercholesterolemia-Induced Oxidative/Nitrative Stress in the Heart. Oxidative Med. Cell. Longev. 2016, 2016, 3863726.
- Alpert, M.A.; Omran, J.; Bostick, B.P. Effects of Obesity on Cardiovascular Hemodynamics, Cardiac Morphology, and Ventricular Function. Curr. Obes. Rep. 2016, 5, 424–434.
- Ebong, I.A.; Goff, D.C.; Rodriguez, C.J.; Chen, H.; Bertoni, A.G. Mechanisms of Heart Failure in Obesity. Obes. Res. Clin. Pract. 2014, 8, e540–e548.
- Opie, L.H.; Commerford, P.J.; Gersh, B.J.; Pfeffer, M.A. Controversies in Ventricular Remodelling. Lancet 2006, 367, 356–367.
- Neeland, I.J.; Gupta, S.; Ayers, C.R.; Turer, A.T.; Rame, J.E.; Das, S.R.; Berry, J.D.; Khera, A.; McGuire, D.K.; Vega, G.L.; et al. Relation of Regional Fat Distribution to Left Ventricular Structure and Function. Circ. Cardiovasc. Imaging 2013, 6, 800–807.
- Liu, J.; Li, J.; Pu, H.; He, W.; Zhou, X.; Tong, N.; Peng, L. Cardiac Remodeling and Subclinical Left Ventricular Dysfunction in Adults with Uncomplicated Obesity: A Cardiovascular Magnetic Resonance Study. Quant. Imaging Med. Surg. 2022, 12, 2035–2050.
- Hammond, I.W.; Devereux, R.B.; Alderman, M.H.; Laragh, J.H. Relation of Blood Pressure and Body Build to Left Ventricular Mass in Normotensive and Hypertensive Employed Adults. J. Am. Coll. Cardiol. 1988, 12, 996–1004.
- Lauer, M.S.; Anderson, K.M.; Kannel, W.B.; Levy, D. The Impact of Obesity on Left Ventricular Mass and Geometry: The Framingham Heart Study. JAMA J. Am. Med. Assoc. 1991, 266, 231–236.
- Jongjirasiri, S.; Waeosak, P.; Laothamatas, J.; Sritara, C.; Saengruang-Orn, S. Effect of Obesity on Left Ventricular Mass: Results from 320 Multi-Slices Computed Tomography. J. Med. Assoc. Thail. 2017, 100, 219–229.
- Bombelli, M.; Facchetti, R.; Sega, R.; Carugo, S.; Fodri, D.; Brambilla, G.; Giannattasio, C.; Grassi, G.; Mancia, G. Impact of Body Mass Index and Waist Circumference on the Long-Term Risk of Diabetes Mellitus, Hypertension, and Cardiac Organ Damage. Hypertension 2011, 58, 1029–1035.
- Lee, T.C.; Jin, Z.; Homma, S.; Nakanishi, K.; Elkind, M.S.V.; Rundek, T.; Tugcu, A.; Matsumoto, K.; Sacco, R.L.; di Tullio, M.R. Changes in Left Ventricular Mass and Geometry in the Older Adults: Role of Body Mass and Central Obesity. J. Am. Soc. Echocardiogr. 2019, 32, 1318–1325.
- Woodiwiss, A.J.; Libhaber, C.D.; Majane, O.H.I.; Libhaber, E.; Maseko, M.; Norton, G.R. Obesity Promotes Left Ventricular Concentric Rather than Eccentric Geometric Remodeling and Hypertrophy Independent of Blood Pressure. Am. J. Hypertens. 2008, 21, 1144–1151.
- Cuspidi, C.; Rescaldani, M.; Sala, C.; Grassi, G. Left-Ventricular Hypertrophy and Obesity: A Systematic Review and Meta-Analysis of Echocardiographic Studies. J. Hypertens. 2014, 32, 16–25.
- Nelson, R.; Antonetti, I.; Bisognano, J.D.; Sloand, J. Obesity-Related Cardiorenal Syndrome. J. Clin. Hypertens. 2010, 12, 59–63.
- Pascual, M.; Pascual, D.A.; Soria, F.; Vicente, T.; Hernández, A.M.; Tébar, F.J.; Valdés, M. Effects of Isolated Obesity on Systolic and Diastolic Left Ventricular Function. Heart 2003, 89, 1152–1156.
- Khan, M.F.; Movahed, M.R. Obesity Cardiomyopathy and Systolic Function: Obesity Is Not Independently Associated with Dilated Cardiomyopathy. Heart Fail. Rev. 2013, 18, 207–217.
- Oh, A.; Okazaki, R.; Sam, F.; Valero-Muñoz, M. Heart Failure With Preserved Ejection Fraction and Adipose Tissue: A Story of Two Tales. Front. Cardiovasc. Med. 2019, 6, 110.
- Wong, C.Y.; O’Moore-Sullivan, T.; Leano, R.; Hukins, C.; Jenkins, C.; Marwick, T.H. Association of Subclinical Right Ventricular Dysfunction with Obesity. J. Am. Coll. Cardiol. 2006, 47, 611–616.
- Krebber, M.M.; van Dijk, C.G.M.; Vernooij, R.W.M.; Brandt, M.M.; Emter, C.A.; Rau, C.D.; Fledderus, J.O.; Duncker, D.J.; Verhaar, M.C.; Cheng, C.; et al. Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Extracellular Matrix Remodeling during Left Ventricular Diastolic Dysfunction and Heart Failure with Preserved Ejection Fraction: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2020, 21, 6742.
- Linssen, P.B.C.; Brunner-La Rocca, H.P.; Schalkwijk, C.G.; Beulens, J.W.J.; Elders, P.J.M.; van der Heijden, A.A.; Slieker, R.C.; Stehouwer, C.D.A.; Henry, R.M.A. Serum Matrix Metalloproteinases and Left Atrial Remodeling—The Hoorn Study. Int. J. Mol. Sci. 2020, 21, 4944.
- Stritzke, J.; Markus, M.R.P.; Duderstadt, S.; Lieb, W.; Luchner, A.; Döring, A.; Keil, U.; Hense, H.W.; Schunkert, H. The Aging Process of the Heart: Obesity Is the Main Risk Factor for Left Atrial Enlargement During Aging. The MONICA/KORA (Monitoring of Trends and Determinations in Cardiovascular Disease/Cooperative Research in the Region of Augsburg) Study. J. Am. Coll. Cardiol. 2009, 54, 1982–1989.
- Heeneman, S.; Cleutjens, J.P.; Faber, B.C.; Creemers, E.E.; van Suylen, R.J.; Lutgens, E.; Cleutjens, K.B.; Daemen, M.J. The Dynamic Extracellular Matrix: Intervention Strategies during Heart Failure and Atherosclerosis. J. Pathol. 2003, 200, 516–525.
- Chen, S.W.; Tung, Y.C.; Jung, S.M.; Chu, Y.; Lin, P.J.; Kao, W.W.Y.; Chu, P.H. Lumican-Null Mice Are Susceptible to Aging and Isoproterenol-Induced Myocardial Fibrosis. Biochem. Biophys. Res. Commun. 2017, 482, 1304–1311.
- Weber, K.T.; Sun, Y.; Tyagi, S.C.; Cleutjens, J.P.M. Collagen Network of the Myocardium: Function, Structural Remodeling and Regulatory Mechanisms. J. Mol. Cell. Cardiol. 1994, 26, 279–292.
- Chute, M.; Aujla, P.; Jana, S.; Kassiri, Z. The Non-Fibrillar Side of Fibrosis: Contribution of the Basement Membrane, Proteoglycans, and Glycoproteins to Myocardial Fibrosis. J. Cardiovasc. Dev. Dis. 2019, 6, 35.
- Spinale, F.G. Myocardial Matrix Remodeling and the Matrix Metalloproteinases: Influence on Cardiac Form and Function. Physiol. Rev. 2007, 87, 1285–1342.
- Fedak, P.W.M.; Verma, S.; Weisel, R.D.; Li, R.-K. Cardiac Remodeling and Failure. Cardiovasc. Pathol. 2005, 14, 49–60.
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146.
- Fan, D.; Takawale, A.; Lee, J.; Kassiri, Z. Cardiac Fibroblasts, Fibrosis and Extracellular Matrix Remodeling in Heart Disease. Fibrogenesis Tissue Repair 2012, 5, 15.
- Ju, H.; Dixon, I.M.C. Cardiac Extracellular Matrix and Its Role in the Development of Heart Failure. In Mechanisms of Heart Failure; Springer: Boston, MA, USA, 1995.
- Miles, C.; Westaby, J.; Ster, I.C.; Asimaki, A.; Boardman, P.; Joshi, A.; Papadakis, M.; Sharma, S.; Behr, E.R.; Sheppard, M.N. Morphometric Characterization of Collagen and Fat in Normal Ventricular Myocardium. Cardiovasc. Pathol. 2020, 48, 107224.
- Prockop, D.J.; Kivirikko, K.I. Collagens: Molecular Biology, Diseases, and Potentials for Therapy. Annu. Rev. Biochem. 1995, 64, 403–434.
- Segura, A.M.; Frazier, O.H.; Buja, L.M. Fibrosis and Heart Failure. Heart Fail. Rev. 2014, 19, 173–185.
- Frangogiannis, N.G. Cardiac Fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488.
- Spinale, F.G.; Gunasinghe, H.; Sprunger, P.D.; Baskin, J.M.; Bradham, W.C. Extracellular Degradative Pathways in Myocardial Remodeling and Progression to Heart Failure. Proc. J. Card. Fail. 2002, 8, S332–S338.
- Heymans, S.; Schroen, B.; Vermeersch, P.; Milting, H.; Gao, F.; Kassner, A.; Gillijns, H.; Herijgers, P.; Flameng, W.; Carmeliet, P.; et al. Increased Cardiac Expression of Tissue Inhibitor of Metalloproteinase-1 and Tissue Inhibitor of Metalloproteinase-2 Is Related to Cardiac Fibrosis and Dysfunction in the Chronic Pressure-Overloaded Human Heart. Circulation 2005, 112, 1136–1144.
- Brower, G.L.; Gardner, J.D.; Forman, M.F.; Murray, D.B.; Voloshenyuk, T.; Levick, S.P.; Janicki, J.S. The Relationship between Myocardial Extracellular Matrix Remodeling and Ventricular Function. Eur. J. Cardio-Thorac. Surg. 2006, 30, 604–610.
- Badenhorst, D.; Maseko, M.; Tsotetsi, O.J.; Naidoo, A.; Brooksbank, R.; Norton, G.R.; Woodiwiss, A.J. Cross-Linking Influences the Impact of Quantitative Changes in Myocardial Collagen on Cardiac Stiffness and Remodelling in Hypertension in Rats. Cardiovasc. Res. 2003, 57, 632–641.
- Norton, G.R.; Tsotetsi, J.; Trifunovic, B.; Hartford, C.; Candy, G.P.; Woodiwiss, A.J. Myocardial Stiffness Is Attributed to Alterations in Cross-Linked Collagen Rather than Total Collagen or Phenotypes in Spontaneously Hypertensive Rats. Circulation 1997, 96, 1991–1998.
- Woodiwiss, A.J.; Tsotetsi, O.J.; Sprott, S.; Lancaster, E.J.; Mela, T.; Chung, E.S.; Meyer, T.E.; Norton, G.R. Reduction in Myocardial Collagen Cross-Linking Parallels Left Ventricular Dilatation in Rat Models of Systolic Chamber Dysfunction. Circulation 2001, 103, 155–160.
- Iimoto, D.S.; Covell, J.W.; Harper, E. Increase in Cross-Linking of Type I and Type III Collagens Associated with Volume-Overload Hypertrophy. Circ. Res. 1988, 63, 399–408.
- Herrmann, K.L.; McCulloch, A.D.; Omens, J.H. Glycated Collagen Cross-Linking Alters Cardiac Mechanics in Volume-Overload Hypertrophy. Am. J. Physiol.-Heart Circ. Physiol. 2003, 284, H1277–H1284.
- Avendano, G.F.; Agarwal, R.K.; Bashey, R.I.; Lyons, M.M.; Soni, B.J.; Jyothirmayi, G.N.; Regan, T.J. Effects of Glucose Intolerance on Myocardial Function and Collagen-Linked Glycation. Diabetes 1999, 48, 1443–1447.
- Liu, J.; Masurekar, M.R.; Vatner, D.E.; Jyothirmayi, G.N.; Regan, T.J.; Vatner, S.F.; Meggs, L.G.; Malhotra, A. Glycation End-Product Cross-Link Breaker Reduces Collagen and Improves Cardiac Function in Aging Diabetic Heart. Am. J. Physiol.-Heart Circ. Physiol. 2003, 285, H2587–H2591.
- Asif, M.; Egan, J.; Vasan, S.; Jyothirmayi, G.N.; Masurekar, M.R.; Lopez, S.; Williams, C.; Torres, R.L.; Wagle, D.; Ulrich, P.; et al. An Advanced Glycation Endproduct Cross-Link Breaker Can Reverse Age-Related Increases in Myocardial Stiffness. Proc. Natl. Acad. Sci. USA 2000, 97, 2809–2813.
- Yamamoto, K.; Masuyama, T.; Sakata, Y.; Nishikawa, N.; Mano, T.; Yoshida, J.; Miwa, T.; Sugawara, M.; Yamaguchi, Y.; Ookawara, T.; et al. Myocardial Stiffness Is Determined by Ventricular Fibrosis, but Not by Compensatory or Excessive Hypertrophy in Hypertensive Heart. Cardiovasc. Res. 2002, 55, 76–82.
- Burgess, M.L.; Buggy, J.; Price, R.L.; Abel, F.L.; Terracio, L.; Samarel, A.M.; Borg, T.K. Exercise- and Hypertension-Induced Collagen Changes Are Related to Left Ventricular Function in Rat Hearts. Am. J. Physiol.-Heart Circ. Physiol. 1996, 270, H151–H159.
- Mukherjee, D.; Sen, S. Collagen Phenotypes during Development and Regression of Myocardial Hypertrophy in Spontaneously Hypertensive Rats. Circ. Res. 1990, 67, 1474–1480.
- Marijianowski, M.M.H.; Teeling, P.; Mann, J.; Becker, A.E. Dilated Cardiomyopathy Is Associated with an Increase in the Type I/Type III Collagen Ratio: A Quantitative Assessment. J. Am. Coll. Cardiol. 1995, 25, 1263–1272.
- Eid, R.A.; Alkhateeb, M.A.; El-Kott, A.F.; Eleawa, S.M.; Zaki, M.S.A.; Alaboodi, S.A.; Salem Al-Shudiefat, A.A.R.; Aldera, H.; Alnamar, N.M.; Alassiri, M.; et al. A High-Fat Diet Rich in Corn Oil Induces Cardiac Fibrosis in Rats by Activating JAK2/STAT3 and Subsequent Activation of ANG II/TGF-1β/Smad3 Pathway: The Role of ROS and IL-6 Trans-Signaling. J. Food Biochem. 2019, 43, e12952.
- Nachar, W.; Merlet, N.; Maafi, F.; Shi, Y.; Mihalache-Avram, T.; Mecteau, M.; Ferron, M.; Rhéaume, E.; Tardif, J.C. Cardiac Inflammation and Diastolic Dysfunction in Hypercholesterolemic Rabbits. PLoS ONE 2019, 14, e0220707.
- Wei, S.; Chow, L.T.C.; Shum, I.O.L.; Qin, L.; Sanderson, J.E. Left and Right Ventricular Collagen Type I/III Ratios and Remodeling Post-Myocardial Infarction. J. Card. Fail. 1999, 5, 117–126.
- Shimizu, M.; Umeda, K.; Sugihara, N.; Yoshio, H.; Ino, H.; Takeda, R.; Okada, Y.; Nakanishi, I. Collagen Remodelling in Myocardia of Patients with Diabetes. J. Clin. Pathol. 1993, 46, 32–36.
- Nagase, H.; Woessner, J.F. Matrix Metalloproteinases. J. Biol. Chem. 1999, 274, 21491–21494.
- Itoh, Y. Membrane-Type Matrix Metalloproteinases: Their Functions and Regulations. Matrix Biol. 2015, 44–46, 44–46.
- Steffensen, B.; Wallon, U.M.; Overall, C.M. Extracellular Matrix Binding Properties of Recombinant Fibronectin Type II-like Modules of Human 72-KDa Gelatinase/Type IV Collagenase: High Affinity Binding to Native Type I Collagen but Not Native Type IV Collagen. J. Biol. Chem. 1995, 270, 11555–11566.
- Gaffney, J.; Solomonov, I.; Zehorai, E.; Sagi, I. Multilevel Regulation of Matrix Metalloproteinases in Tissue Homeostasis Indicates Their Molecular Specificity In Vivo. Matrix Biol. 2015, 44–46, 44–46.
- Visse, R.; Nagase, H. Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases: Structure, Function, and Biochemistry. Circ. Res. 2003, 92, 827–839.
- Fedak, P.W.M.; Altamentova, S.M.; Weisel, R.D.; Nili, N.; Ohno, N.; Verma, S.; Lee, T.Y.J.; Kiani, C.; Mickle, D.A.G.; Strauss, B.H.; et al. Matrix Remodeling in Experimental and Human Heart Failure: A Possible Regulatory Role for TIMP-3. Am. J. Physiol.-Heart Circ. Physiol. 2003, 284, H626–H634.
- Lambert, E.; Dassé, E.; Haye, B.; Petitfrère, E. TIMPs as Multifacial Proteins. Crit. Rev. Oncol./Hematol. 2004, 49, 187–198.
- Timms, P.M.L.; Wright, A.; Maxwell, P.; Campbell, S.; Dawnay, A.B.; Srikanthan, V. Plasma Tissue Inhibitor of Metalloproteinase-1 Levels Are Elevated in Essential Hypertension and Related to Left Ventricular Hypertrophy. Am. J. Hypertens. 2002, 15, 269–272.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
813
Entry Collection:
Hypertension and Cardiovascular Diseases
Revisions:
2 times
(View History)
Update Date:
26 Apr 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No