Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Huang, Y.; , . Induction of Accelerated Aging in a Mouse Model. Encyclopedia. Available online: https://encyclopedia.pub/entry/22270 (accessed on 26 July 2026).
Huang Y, . Induction of Accelerated Aging in a Mouse Model. Encyclopedia. Available at: https://encyclopedia.pub/entry/22270. Accessed July 26, 2026.
Huang, Yan, . "Induction of Accelerated Aging in a Mouse Model" Encyclopedia, https://encyclopedia.pub/entry/22270 (accessed July 26, 2026).
Huang, Y., & , . (2022, April 26). Induction of Accelerated Aging in a Mouse Model. In Encyclopedia. https://encyclopedia.pub/entry/22270
Huang, Yan and . "Induction of Accelerated Aging in a Mouse Model." Encyclopedia. Web. 26 April, 2022.
Copy Citation
With the global increase of the elderly population, the improvement of the treatment for various aging-related diseases and the extension of a healthy lifespan have become some of the most important current medical issues. In order to understand the developmental mechanisms of aging and aging-related disorders, animal models are essential to conduct relevant studies. Among them, mice have become one of the most prevalently used model animals for aging-related studies due to their high similarity to humans in terms of genetic background and physiological structure, as well as their short lifespan and ease of reproduction.
aging
aging-related disease
mouse model
1. Introduction
The world’s population continues to grow, with those over 65 being the fastest growing age group. In 2019, one out of every eleven people in the worldwide population was over the age of 65. By 2050, this proportion will increase to one in every six people over the age of 65. In some regions, such as in Europe and North America, one in four people will be over 65 years old. Aging is a key risk factor for multiple disorders. More than 75% of the elderly suffer from at least one chronic disease, and the problem of “unhealthy longevity” for the elderly is prominent [1][2].
As countries face the challenges of aging populations, there is a need to promote healthy aging and provide adequate social protection. To promote a healthy aging process and prevent aging-related health problems, correctly understanding aging mechanisms and developing effective and affordable intervention strategies for anti-aging have great social significance and huge economic benefits. During this process, an animal model of aging is a powerful tool for researchers to study the mechanism of aging.
Generally, aging models are divided into two categories: natural aging models and accelerated aging models. In the process of aging, naturally aging mice develop many phenotypes similar to normal human aging like cataracts [3] and muscle weakness [4]. For example, the most well-known strain of mice, C57BL/6, have a lifespan of two to three years, while naked mole-rats can live up to 30 years. One longevity mechanism of naked mole-rats is their effective DNA damage repair system. However, research on natural aging model is time-consuming, labor-intensive, and expensive, with large individual variations compared to the accelerated aging models [5][6]. As such, the induced accelerated aging model is favored by researchers because of its convenient source, short modeling time, and relatively controllable aging effect. For aging and aging-related chronic disorders, mice have become one of the most important animal models for studying various aging-related human diseases due to their similarities to humans in terms of genetic background and the structure and function of various organs or systems, as well as the advantages of short lifespan and ease of reproduction [6][7]. With the help of mouse models, researchers have revealed the mechanisms of aging as well as the pathogenesis of various chronic diseases, and many therapeutic approaches for such chronic diseases have been validated in mouse models preclinically.
2. Systemic-Induced Accelerated Aging Mouse Model
In this section, researchers will review some commonly used mouse models of accelerated systemic aging including drug treatment, genetic engineered models, irradiation induction, etc. They are characterized by an aging phenotype in multiple tissues or organs, reflecting systemic aging. The systemic-induced accelerated aging mouse models are summarized in Table 1.
Table 1. Summary of systemic-induced accelerated aging mouse models.
| Type | Subdivision | Phenotypes |
|---|---|---|
| D-galactose-induced senescence model | Brain | Cognitive impairment Mitochondrial dysfunction Neuronal degeneration Apoptosis Depressive and anxious |
| Heart | Cardiac fibrosis Collagen accumulation Fibroblasts disordered arrangement |
|
| Kidney | Kidney index ↓ Uric acid & Cys-C ↑ Glomerular and tubular damage ↑ |
|
| Liver | Liver fibrosis Glycogen levels ↓ Lipid deposition ↑ |
|
| Reproductive system |
Estrogen and progesterone ↓ Ovarian follicle regression Uterine wall endometrial gland atrophy Disrupt estrous cycles |
|
| Intestinal flora | Disturbance | |
| Lung | Oxidative stress ↑ Fibrotic status Chronic inflammation |
|
| SAMP mice | SAMP 1 | Aging amyloidosis Immune dysfunction Renal atrophy Hearing loss Senile pulmonary hyperinflation |
| SAMP 6 | Senile osteoporosis Myeloid progenitor cell senescence |
|
| SAMP 8 | Astrogliosis Microgliosis Neurodegeneration Amyloid accumulation MAPT hyperphosphorylation |
|
| SAMP 10 | Learning and memory impairment Cerebral cortex and limbic system atrophy |
|
| Rps9 D95N mouse | Altered fur Cataracts Hunched posture Body composition function & body weight ↓ Fat mass & muscle strength ↓ Shortened lifespan Mouse urinary syndrome Extramedullary hematopoiesis |
|
| Lama−/− | Short lifespan Growth retardation Muscular dystrophy Altered lipid metabolism |
|
| Wrn∆hel/∆hel | Short lifespan Abnormal hyaluronic acid excretion Metabolic abnormalities Increased genomic instability and cancer incidence |
|
| Csa−/−, Csb−/− | Short lifespan Reduced fat mass Photoreceptor cell loss Neural pathology |
|
| Progeria syndrome mouse | XpdTTD/TTD | Short lifespan Trichothiodystrophy |
| Bub1bH/H, Xpg−/− | Brain atrophy Neuronal loss Neurofibrillary deposition of Aβ or senile plaques |
|
| Bub1bH/H, Bub1b+/GTTA | Mean muscle fiber diameter ↓ Muscle fiber size variation ↑ Intermuscular fibrosis Regenerative capacity of skeletal muscles ↓ |
|
| Mitochondrial DNA polymerase mutant mouse | Lifespan ↓ Weight loss Subcutaneous fat ↓ Hair loss Kyphosis Osteoporosis Anemia Fertility ↓ Spermatogonia depletion Heart enlargement |
|
| Total body irradiation (TBI) model | Progressive premature frailty Cognitive decline Whole blood antioxidant capacity ↓ RBC glutathione ↓ Thymic involution Articular cartilage and bone degeneration Ovarian environment damage |
|
| Ozone-induced senescence model | Cognitive decline Memory impairment AD symptoms Lung tumor growth ↑ |
|
| Chronic jet-lag mouse | Accelerated initial tumor growth Shortened mouse survival Induce spontaneous hepatocellular carcinoma Obesity Depression Addiction Abnormal cardiac structure Impaired cardiac function |
2.1. The D-Galactose-Induced Senescence Model
D-galactose is a common aldohexose that exists naturally in the body and in daily foods [8]. After ingestion, a healthy adult can metabolize and eliminate a maximum daily dose of 50 g of galactose from the body within about eight hours [9]. However, when galactose accumulates to high levels, reactive oxygen species (ROS) are generated by mitochondrial respiratory chain enzymes, xanthine oxidase, lipoxygenase, cyclooxygenase, nitric oxide synthase, and peroxidase. Increased ROS can subsequently lead to elevated oxidative stress and inflammation, inducing mitochondrial dysfunction and apoptosis [10]. Meanwhile, the elevated mitochondrial ROS level can lead to the activation of many biochemical pathways, such as the polyol pathway, the formation of advanced glycation end products (AGEs), the activation of protein kinase C, and the hexosamine pathway [11][12]. Overall, D-galactose-induced methods can increase aging markers such as AGEs, receptors for advanced glycation end products (RAGEs), aldose reductase (AR), sorbitol dehydrogenase (SDH), decreased telomerase activity, shortened telomere, β-site amyloid precursor protein cleaving enzyme 1 (BACE-1), amyloid β (Aβ), aging-related pathways (p16, p21, p53, etc.), and positive senescence-associated β-galactosidase (SA-β-gal) staining [13]. Multiple tissues and organs, including the brain, heart, lung, liver, kidney, reproductive system, gastrointestinal system, and so on, manifest aging phenotypes after D-galactose treatment [14].
Previous works have shown that D-galactose induces brain aging by increasing mitochondrial dysfunction, oxidative stress, inflammation, apoptosis, and decreasing the expression of brain-derived neurotrophic factor. D-galactose injections may induce brain aging similar to human brain aging in many ways, including mitochondrial dysfunction, increased oxidative stress, decreased ATP production, neuronal degeneration and apoptosis, and cognitive deficits [13][15][16][17]. D-galactose increases the neuro-inflammation markers via activating NF-κB, leading to memory impairment [18][19]. Besides learning and memory inhibition, D-galactose-treated mice also exhibit depressive and anxious behaviors [20].
The leading cause of death in elderly people worldwide is cardiovascular disease [21]. D-galactose treatment increases the risk of cardiovascular disease, which is associated with excess ROS and oxidative stress. Persistent oxidative stress has been revealed to be related to decreased ferric reducing antioxidant power and lower activity of Cu-Zn superoxide dismutase, leading to myocardial damage [22]. Studies have shown that galactose reduces endogenous hydrogen sulphide producing enzyme cystathionine γ-lyase (CSE) [23] and antioxidant enzymes such as catalase (CAT), haem oxygenase-1 (HO-1), superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), and nitric oxide synthase (NOS), leading to decreased total antioxidant capacity and inducing lipid peroxidation markers including malondialdehyde (MDA), lipid hydroperoxides (L-OOH), and conjugated dienes (CD) in cardiac tissue [24][25]. D-galactose increases whole heart weight and left ventricle weight, which is associated with hypertension and aging. At the same time, the heart tissue showed enlarged myocardial fibers, blurred structure, shortened distortion, widening of the interval, and obvious capillaries of myocardial interstitial congestion [26]. D-galactose treatment resulted in cardiac fibrosis, significant accumulated collagen, and disordered arrangement of fibroblasts compared with the control. D-galactose also increased cardiac apoptosis markers [27]. Excessive D-galactose can be transformed to advanced AGEs via the Maillard reaction [28]. AGEs bind to the receptors, RAGE, increasing ROS production via NADPH oxidase. NADPH oxidase further activates p38 MAP kinases, causing transcription factor NF-κB translocating into the nucleus, where transcription of inflammatory cascades like tumor necrosis factor alpha (TNF-α) are enhanced [29]. D-galactose treatment can also increase fibrotic markers such as connective tissue growth factor (CTGF), transforming growth factor β1 (TGF-β1), phosphorylated mitogen-activated protein kinase 1/2 (p-MEK1/2), phosphorylated extracellular signal-regulated kinase 1/2 (p-ERK1/2), matrix metalloproteinase (MMP), and pathological specific protein 1 (SP1) [30].
D-galactose treatment also increases oxidative markers (e.g., MDA) and decreases antioxidant enzymes (e.g., SOD, GSH-Px and NOS) in lung, liver, and renal tissues [26][31][32][33][34][35]. D-galactose treatment can affect lung elastic constitution. The primary effects of treatment on the lungs are increased alveolar size and reduced elastic recoil, which may facilitate airway closure [36]. D-galactose treatment is considered to successfully mimic the natural aging process by increasing oxidative stress, fibrotic status, and chronic inflammation in the lungs.
The liver is the main site where D-galactose is metabolized, thus excess D-galactose in the body may significantly affect the liver. As previously mentioned, high levels of D-galactose can react with free amines in the amino acids to form AGEs, which is found to be involved in the progression of various liver diseases [12]. High levels of D-galactose lead to the accumulation of its final metabolite, galactitol, which will eventually lead to ROS accumulation through the p38 MAPK/NRF2/HO-1 signaling pathway and cause cellular osmotic stress in the liver [37]. D-galactose treatment significantly increased apoptotic proteins including Bax, procaspase-3, and caspase-3, and raised the ratio of Bax/Bcl-2 in the liver tissue [34][38][39]. Various changes similar to natural aging were also observed in the D-galactose-treated livers. Compared with controls, the livers of D-galactose-treated animals had lower levels of glycogen and more lipid deposition. Masson’s trichrome staining showed obvious collagen fibers around blood vessels and in the interphase of liver tissue, and irregular pseudo-lobules around liver tissue, indicating a state of liver fibrosis [40][41].
D-galactose treatment significantly reduced the renal index of animals, and markers of acute kidney injury such as uric acid and cystatin C (Cys-C) were also increased [42]. In the kidney, D-galactose administration resulted in an increase in the TUNEL-positive cells and DNA fragmentation. In addition, p21 expression and the staining intensity of SA-β-gal were also increased in kidney cells [43]. Extensive glomerular and tubular damages were detected in D-galactose-treated animals, as the number of tubules with cellular necrosis from the renal cortices and outer medulla were significantly increased [43][44].
In the male reproductive system, D-galactose induced oxidative stress, marked by an increase in MDA levels in the prostate, testis, epididymis, and decrease in SOD activity in the testis. Peroxidation in the testicular and epididymal mitochondrial fractions was also significantly increased after D-galactose treatment [45]. Female reproductive aging is characterized by decreased levels of estrogen, progesterone, inhibin B, anti-Müllerian hormone, and androgens, which include free testosterone, dehydroepiandrosterone (DHEAS), and androstenedione [46]. Compared to the control groups, D-galactose administration produced aging-associated changes like reduced estrogen and progesterone levels and increased FSH and luteinizing hormone (LH) levels. Ovarian follicle regression and atrophy on the uterine wall and endometrial gland were observed after D-galactose treatment, which indicates disrupted estrous cycles and damaged uterine and ovarian tissues [47].
D-galactose injection can lead to changes in the level of oxidative stress that affect the microbial environment in the intestine and lead to intestinal flora disorder [48]. The ecology of intestinal flora is closely related to aging, and intestinal ecological disturbance can lead to accelerated aging and a shortened lifespan [49]. Transferring gut microbiota from aged to young germ-free mice triggered innate immune and inflammatory responses. Effects of aging include increased differentiation of CD4+ T cells in the spleen, upregulation of intestinal inflammatory cytokine such as TNF-α, and increased cycling of bacterial-derived inflammatory cytokines [50][51]. In addition, D-galactose administration significantly decreased the small intestine propulsion rates and prolonged gastrointestinal transit time [49]. The aging gut triggers chronic inflammation, leading to gut dysplasia and intestinal dysplasia in turn leads to defective epithelial function, predisposing the host to infection, neoplasia, and increased mortality [52].
2.2. Senescence-Accelerated Mouse/Prone
Researchers from Kyoto University found an aging phenotype from a subset of pups while maintaining an inbred line of AKR/J mice. Characteristics of these mice include hair loss, reduced activity, and a shortened lifespan. These aging traits are thought to develop as a result of elevated oxidative stress and are inherited by their offspring. Further, the accelerated aging mice were grouped into several distinct subtypes according to their phenotypes [53].
Senescence-accelerated mouse/prone (SAMP) is a group of inbred mouse strains that typically exhibit accelerated aging [54]. Meanwhile, since it shows various aging-related diseases similar to humans, such as aging amyloidosis, senile osteoporosis, learning/memory impairment, etc., in specific lines, it is widely used for aging research. Cellular senescence in various cell types, including astrocytes, endothelial cells, progenitor cells, retinal epithelial cells, and fibroblasts, was found in the aging SAMP mice [55][56][57]. SAMP mice exhibit an increase in ROS generated by mitochondria or other cellular sites, which not only causes damage to mitochondria, but also triggers degradation that leads to the aging outcome [58].
SAMP1 mice are characterized by aging amyloidosis, immune dysfunction, renal atrophy, hearing loss, and senile pulmonary hyperinflation [59][60]. The lifespan of the SAMP1 mice is about 40% shorter than senescence-accelerated resistant mice (SAMR1), and various signs of aging appear early in appearance.
The SAMP6 mice model is a senile osteoporosis model animal that tends to develop osteoporosis at an early stage with aging due to its low bone density in instar [61]. Bone marrow transplantation experiments have revealed that the cause of SAMP6 osteoporosis is abnormalities in bone marrow stem cells [62]. The incidence of spontaneous leg fractures due to osteoporosis is high in adult SAMP6 mice. Cellular senescence in myeloid progenitors disrupts their differentiation in favor of adipogenesis over osteogenesis. This mechanism is thought to contribute to low osteoblast activity and osteoporosis in SAMP6 mice and the elderly [61][63][64].
The SAMP8 mice develop age-associated deficits in learning and memory and also exhibit various age-related neuropathological changes similar to aging humans [65][66]. Neuropathological changes including astrogliosis, microgliosis, and neurodegeneration occur as early as five months of age [67]. SAMP8 mice also showed accumulated amyloid and age-related microtubule-associated protein tau (MAPT) hyperphosphorylation, as well as increased nitric oxide synthase activity, further demonstrating their feasibility as a brain aging model [68][69]. In addition, decreased activities in SOD, CAT, glutathione reductase, and GSH-Px, and increased activity in acyl-CoA oxidase were detected in SAMP8 mice at 1–12 months of age [70]. SAMP6 and SAMP8 mice also develop many other age-related diseases, including retinal degeneration, testosterone deficiency [71], myocardial fibrosis [72], and hepatic lipid deposition [73].
The SAMP10 mice exhibit learning and memory impairment with aging, and atrophy is observed in the cerebral cortex and limbic system, so it is considered to be a spontaneous model animal for aging and brain degeneration. The atrophy of the frontal cerebral cortex and olfactory bulb is marked in SAMP10 mice [74][75].
So far, more than ten strains of SAMP mice have been identified and widely used in aging studies, each of which can develop various age-related diseases such as renal fibrosis (shrinking of the kidneys), immune dysfunction, and degenerative joint disease like osteoarthritis (OA), etc.
2.3. Rps9 D95N Mouse
Rps9 D95N is a ribosome ambiguity mutation that causes error-prone protein synthesis in mammalian ribosomes, resulting in increased error-prone translation. Rps9 D95N mutant mice exhibit features of accelerated aging, including morphological (altered fur, cataracts, and hunched posture), physiological (body composition and function, body weight, fat mass, and muscle strength), and pathological (shortened lifespan, mouse urinary syndrome and extramedullary hematopoiesis), which also complements and explains the link between accumulation of erroneous proteins resulting from protein mistranslation and individual aging [76].
2.4. Progeria Syndrome Mouse
Mouse models of progeria syndrome have emerged as an attractive tool for evaluating intervention strategies for unhealthy aging due to their short lifespan, relatively simple strategies by single gene deletion or mutation, and their strong phenotypic similarities to normal aging [7]. To understand important mechanisms of the aging process, progeria mice (Lmna−/−, Wrn∆hel/∆hel, Csa−/− or Csb−/−) from common progeria types such as Hutchinson-Gilford Progeria, Werner Syndrome, and Cockayne Syndrome are being widely used.
The most common phenotypes in mouse models of progeria can be observed in bones, joints, skin, nervous system, adipose tissue, skeletal muscle, cardiovascular system, liver, kidney, and the hematopoietic system. Less common lesions occur in the gonads, eyes, and occasionally in the gastrointestinal tract [77].
Some human progeria syndromes like Werner Syndrome exhibit osteoporosis. Current data from a mouse model of progeria indicate that senile osteoporosis is the result of reduced bone turnover and loss of bone mass due to defects in osteoblast progenitor cells, osteoblast differentiation, or osteoblast function [77]. Degenerative joint disease is another major symptom that affects the elderly. Mutation in the Xpd gene of nucleotide excision repair (NER) leads to a short lifespan, causing trichothiodystrophy (TTD) in humans. As expected, XpdTTD/TTD mice exhibited a significant decrease in subchondral bone plate thickness compared to that observed in wild-type mice. Surprisingly, female XpdTTD/TTD mice exhibited less cartilage damage and fewer lost articular cartilage, compared to WT females [78].
During the aging process, skeletal muscle was inevitably accompanied with a reduction in muscle mass, known as sarcopenia or age-related muscle atrophy, and macroscopic examination of sarcopenic mice will show weight loss and marked reduction in muscle mass. Sarcopenia is characterized by the loss of muscle fibers and smaller fiber cross-sectional area that is defined as fiber atrophy [79]. The progeria mouse model Bub1bH/H and Bub1b+/GTTA mice showed decreased mean muscle fiber diameter, increased myofiber size variation, increased intermuscular fibrosis, and impaired regenerative capacity in skeletal muscles [80].
Common brain aging diseases include Alzheimer’s disease (AD) and cognitive dysfunction syndrome. Brain atrophy, neuronal loss, neurofibrillary deposition of Aβ or senile plaques, intraneuronal tauopathies (neurofibrillary tangles, NFTs), cerebrovascular amyloid angiopathy, neuronal lipofuscinosis, vascular and meningeal calcification, decreased white matter integrity, and astrogliosis are common age-related neurological pathologies [81]. The brains of several mouse models of progeria show neurodegenerative changes at an early stage. Similar to age-related gliosis, Bub1bH/H mice have increased numbers of astrocytes and microglia at one and five months of age, respectively. Additionally, Xpg−/− mice developed more astrocytes and increased activation of microglia in the brain and spinal cord [82].
3. Tissue-, Organ-, or System-Specific Mouse Models of Aging-Related Diseases
Among the leading effects of ageing is the heightened incidence of various aging-related diseases, and mouse models continue to serve an essential part in the study of the pathogenesis and treatment of these illnesses. A variety of commonly used and emerging mouse models have been developed for different aging-related diseases, with the aim of reproducing as closely as possible the progression of the diseases in humans (Figure 1).
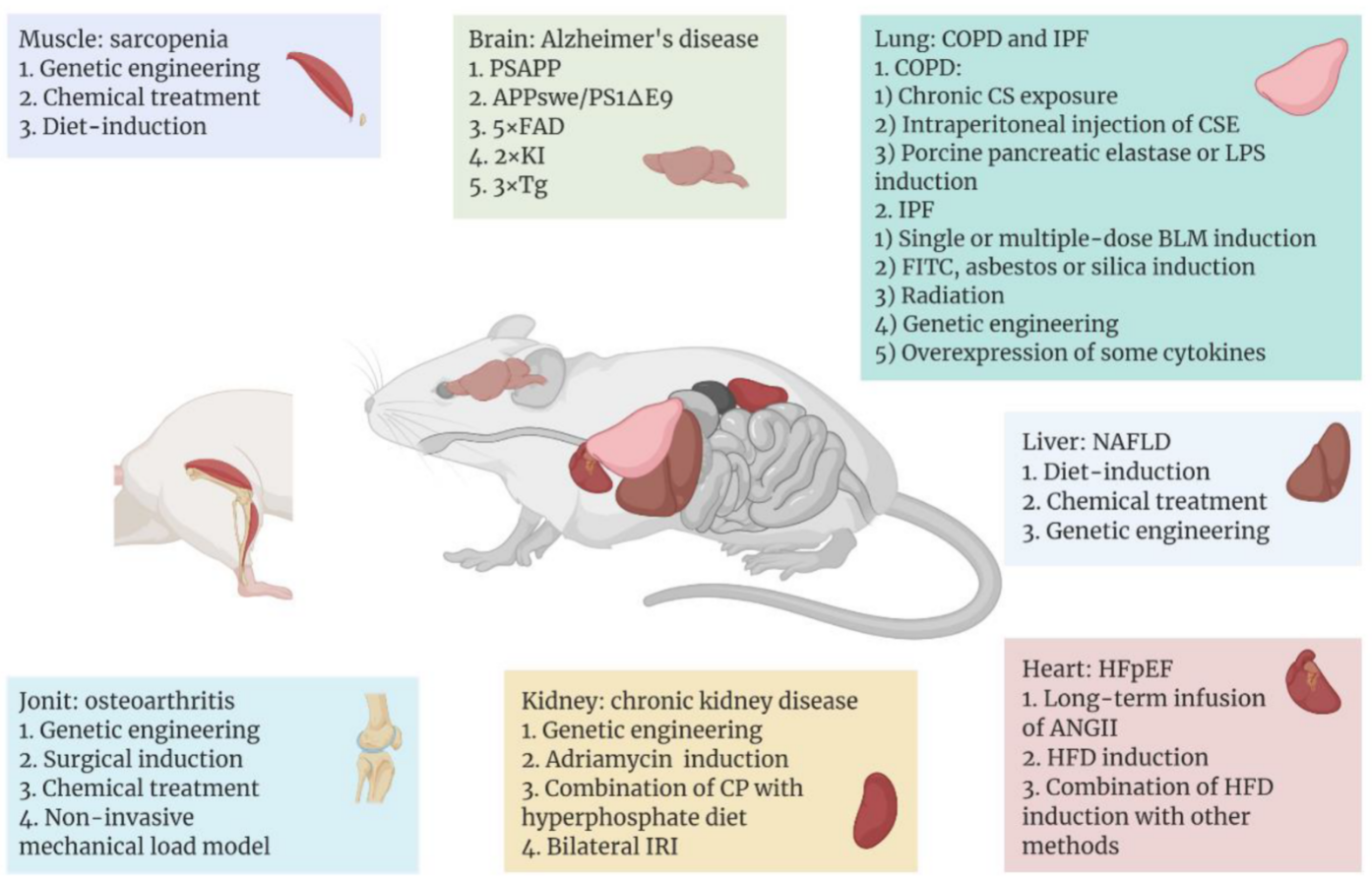
Figure 1. Aging-related diseases include Alzheimer’s disease (AD), sarcopenia, heart failure with preserved ejection fraction (HFpEF), non-alcoholic fatty liver disease (NAFLD), chronic kidney disease (CKD), osteoarthritis, chronic obstructive pulmonary disease (COPD), and idiopathic pulmonary fibrosis (IPF), which occur in different tissues or organs. With the help of mice as model organisms, researchers have used different methods to establish disease models in different tissues or organs of mice. In recent years, some common or emerging methods of modeling aging-related diseases are shown above, and these methods are classified according to the organ to which the disease mainly affects. Figure 1 was created with BioRender software (https://biorender.com/ (accessed on 20 April 2022)).
3.1. Model of Aging Brain or Nerve System
Although there are some genetic similarities between mice and humans, the two still diverge in the expression levels of certain age-related genes, which leads to potentially different aging process in the central nervous system of humans and mice [83]. Therefore, changing the expression levels of those specific genes that contribute to the aging process of the brain is the main strategy for modeling the aging brain in mice. For now, to ensure that experimental results can be extrapolated to humans, research on such models has mainly concentrated on evolutionarily conserved mechanisms that modulate aging. These conserved mechanisms include genomic instability, epigenetic changes, telomere attrition, mitochondrial dysfunction, loss of proteostasis, and dysregulated nutrient sensing [83][84].
For instance, as a neurodegenerative disease, AD represents one of the most common neurological disorders. The number of people affected by the disease has been recorded as over tens of millions worldwide, and the number will continue to rise. Also, it is the most common cause of dementia [85].
So far, the etiology of AD is not yet completely understood. It is thought that genetic elements play key roles in the onset of such disease [86]. The typical histopathological features of AD are Aβ deposits and NFTs in the brain. However, wild-type mice do not spontaneously exhibit symptoms of AD [87]. Because AD-related proteins differ in sequence, pathogenicity, and number of isoforms between rodents and humans [88], the main strategy for modeling AD in mice is to construct transgenic mice that cause amyloid deposition or NFTs in the brain.
Common AD-associated mutant proteins in humans include Aβ, presenilin 1 (PS1), apolipoprotein E (ApoE), and MAPT. In the 1990s, the first transgenic AD model mice stably expressing the mutant human Aβ precursor protein (APP) were constructed [89]. After this, transgenic mice carrying multiple human AD-related mutant proteins emerged. One of the most commonly used AD models today is hAPP/PS1 lines, which carry both mutated human APP and PSEN1, including transgenic strains PSAPP, APPswe/PS1ΔE9, 5XFAD, and 2xKI [90]. Compared to monogenic lines containing only mutated APP or PS1, these transgenic mice exhibit earlier and faster onset of amyloid accumulation and cognitive impairment [91]. However, such AD models do not exhibit signs of NFTs, which can be imitated by mouse models that express human MAPT. Based on this, Oddo et al. constructed a 3xTg model combining the human APP, PS1, and MAPT mutations [92]. Since this model can show both Aβ deposition and NFTs in the brain, it is considered to be the well-established transgenic model of AD. Consistent with patients with AD, some of these transgenic mouse models (e.g., PS19, APPswe/PS1ΔE9) showed increased levels of NF-κB pathway-related proteins (IKKβ, p65, and COX-2) in the brain, indicating upregulation of brain inflammation in these mice. Therefore, the use of such mouse models will also help researchers to further clarify the relationship between neuroinflammation and the pathogenesis of AD [93][94].
3.2. Model of Aging Muscle
The aging of the body is accompanied by the aging of the skeletal muscles. Among other things, sarcopenia, which is a widespread progressive skeletal muscle disorder, is associated with an increased probability of adverse consequences like falls, fractures, physical disability, and death, and its risk increases with age [95]. Modeling of sarcopenia in mice is divided into two main categories: models of genetic engineering and chemical or dietary-induced models.
For genetic engineering models, the majority of research has employed knockout (KO) mice. For example, mitofusion2 (Mfn2) is one of the important protein components mediating mitochondrial fusion, and Mfn2 KO mice exhibit mitochondrial dysfunction in skeletal muscle cells and specific atrophy of type IIb glycolytic fibers [96]. Collagen VI, an extracellular matrix (ECM) protein, has a critical role in skeletal muscle. Six-month-old Col6α1−/− mice exhibit alterations of the diaphragm consistent with aged wild-type mice, such as abnormal tricarboxylic acid (TCA) cycle and decreased autophagy in diaphragm cells, indicating the Col6α1−/− mouse could be considered as a premature model of skeletal muscle aging [97]. Additionally, interleukin 10 (IL-10) [98], SOD1 [99], and NOD-like receptor protein 3 (NLRP3) [100] deficient mice have also been employed in studying the pathogenesis of sarcopenia as well as the intervention of therapeutically targeting such a disease. Overexpression of certain proteins can also lead to sarcopenia, such as TNF-α transgenic mice that exhibit reduced muscle mass, muscle fiber diameter, and Pax7+ muscle stem-cell content [101].
For chemical or diet-induced sarcopenia models, dexamethasone is a common inducer, which is essentially a glucocorticoid that triggers muscle atrophy in mice. It is shown that dexamethasone induces upregulation of ubiquitin ligases in muscle, including muscle atrophy F-box (MAFbx) and muscle ring finger 1 (MuRF1), which may further mediate the degradation of muscle atrophy-associated proteins [102][103]. In addition, diet-induced sarcopenia mouse models allow researchers to investigate the co-occurrence of sarcopenia with other disorders, e.g., sarcopenia can also develop from the reduction in muscle mass and strength caused by certain chronic diseases. Fabián et al. [104] treated mice with hepatotoxin 5-diethoxycarbonyl-1,4-dihydrocollidine (DDC), thereby inducing sarcopenia secondary to chronic liver disease (CLD), as evidenced by reduced muscle strength and motility, as well as the reduction in muscle fiber size and its type of transformation in mice.
3.3. Model of Aging Heart
Heart failure is a complex disease that can eventually result from almost all cardiovascular disorders, like myocardial infarction, atherosclerosis, and hypertension. Based on the left ventricular ejection fraction, heart failure is clinically classified into two major categories: heart failure with reduced ejection fraction or preserved ejection fraction (HFpEF) [105]. Among them, due to the increasing morbidity and mortality of HFpEF in recent years and the lack of effective therapeutic options for this disease, research on HFpEF has received increasing attention and as a result, some mouse models of HFpEF have been developed.
Long-term infusion of angiotensin II (ANGII) into mice based on a mini-osmotic pump is one of the most common methods of modeling HFpEF. Elevated levels of ANGII in the mouse circulatory system can trigger vasoconstriction, hypertension, aldosterone secretion, TGF-β-mediated inflammation, and fibrosis, and ultimately cardiac hypertrophy [105]. These symptoms closely resemble those exhibited by HFpEF in humans. Furthermore, in addition to causing obesity, a high-fat diet (HFD) is also known to induce a host of cardiac-related symptoms, including left ventricular hypertrophy, HFpEF, and diastolic dysfunction [106][107][108][109]. In addition, Withaar et al. [110] constructed a model with HFD and ANGII treatment, which exhibited higher levels of cardiac fibrosis as well as more severe diastolic dysfunction and cardiac hypertrophy compared to the single-treatment group with HFD or ANGII. Also, Schiattarella et al. [111] developed a model in which both HFD and the constitutive NO synthase inhibitor Nω-nitro-L-arginine methyl ester (L-NAME) were imposed. Although L-NAME caused an increase in diastolic and systolic blood pressure, the HFD-L-NAME group exhibited more significant cardiomyocyte hypertrophy and a reduction of myocardial capillary density.
References
- DESA|United Nations. Available online: https://www.un.org/en/desa (accessed on 20 March 2022).
- World Population Prospects 2019. Available online: https://population.un.org/wpp/ (accessed on 20 March 2022).
- Graw, J. Mouse models of cataract. J. Genet. 2009, 88, 469–486.
- Brooks, S.V.; Faulkner, J.A. Skeletal muscle weakness in old age: Underlying mechanisms. Med. Sci. Sports Exerc. 1994, 26, 432–439.
- Gurkar, A.U.; Niedernhofer, L.J. Comparison of mice with accelerated aging caused by distinct mechanisms. Exp. Gerontol. 2015, 68, 43–50.
- Vanhooren, V.; Libert, C. The mouse as a model organism in aging research: Usefulness, pitfalls and possibilities. Ageing Res. Rev. 2013, 12, 8–21.
- Harkema, L.; Youssef, S.A.; de Bruin, A. Pathology of Mouse Models of Accelerated Aging. Vet. Pathol. 2016, 53, 366–389.
- Acosta, P.B.; Gross, K.C. Hidden sources of galactose in the environment. Eur. J. Pediatrics 1995, 154, S87–S92.
- Morava, E. Galactose supplementation in phosphoglucomutase-1 deficiency; review and outlook for a novel treatable CDG. Mol Genet. Metab. 2014, 112, 275–279.
- Ullah, F.; Ali, T.; Ullah, N.; Kim, M.O. Caffeine prevents d-galactose-induced cognitive deficits, oxidative stress, neuroinflammation and neurodegeneration in the adult rat brain. Neurochem. Int. 2015, 90, 114–124.
- Wu, D.M.; Lu, J.; Zheng, Y.L.; Zhou, Z.; Shan, Q.; Ma, D.F. Purple sweet potato color repairs d-galactose-induced spatial learning and memory impairment by regulating the expression of synaptic proteins. Neurobiol. Learn Mem. 2008, 90, 19–27.
- Azman, K.F.; Safdar, A.; Zakaria, R. D-galactose-induced liver aging model: Its underlying mechanisms and potential therapeutic interventions. Exp. Gerontol. 2021, 150, 111372.
- Shwe, T.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. Role of D-galactose-induced brain aging and its potential used for therapeutic interventions. Exp. Gerontol. 2018, 101, 13–36.
- Azman, K.F.; Zakaria, R. D-Galactose-induced accelerated aging model: An overview. Biogerontology 2019, 20, 763–782.
- Li, W.; Wang, S.; Wang, H.; Wang, J.; Jin, F.; Fang, F.; Fang, C. Astragaloside IV Prevents Memory Impairment in D-galactose-induced Aging Rats Via the AGEs/RAGE/ NF-kappaB Axis. Arch. Med. Res. 2022, 53, 20–28.
- Banji, O.J.; Banji, D.; Ch, K. Curcumin and hesperidin improve cognition by suppressing mitochondrial dysfunction and apoptosis induced by D-galactose in rat brain. Food Chem. Toxicol. 2014, 74, 51–59.
- Khedr, N.F.; Werida, R.H.; Abo-Saif, M.A. Candesartan protects against d-galactose induced—Neurotoxicity and memory deficit via modulation of autophagy and oxidative stress. Toxicol. Appl. Pharm. 2022, 435, 115827.
- Long, J.; Wang, X.; Gao, H.; Liu, Z.; Liu, C.; Miao, M.; Cui, X.; Packer, L.; Liu, J. D-galactose toxicity in mice is associated with mitochondrial dysfunction: Protecting effects of mitochondrial nutrient R-alpha-lipoic acid. Biogerontology 2007, 8, 373–381.
- Kumar, A.; Dogra, S.; Prakash, A. Effect of carvedilol on behavioral, mitochondrial dysfunction, and oxidative damage against D-galactose induced senescence in mice. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2009, 380, 431–441.
- Haider, S.; Liaquat, L.; Shahzad, S.; Sadir, S.; Madiha, S.; Batool, Z.; Tabassum, S.; Saleem, S.; Naqvi, F.; Perveen, T. A high dose of short term exogenous D-galactose administration in young male rats produces symptoms simulating the natural aging process. Life Sci. 2015, 124, 110–119.
- Sacco, R.L.; Roth, G.A.; Reddy, K.S.; Arnett, D.K.; Bonita, R.; Gaziano, T.A.; Heidenreich, P.A.; Huffman, M.D.; Mayosi, B.M.; Mendis, S.; et al. The Heart of 25 by 25: Achieving the Goal of Reducing Global and Regional Premature Deaths from Cardiovascular Diseases and Stroke: A Modeling Study from the American Heart Association and World Heart Federation. Circulation 2016, 133, e674–e690.
- Cebe, T.; Yanar, K.; Atukeren, P.; Ozan, T.; Kuruç, A.I.; Kunbaz, A.; Sitar, M.E.; Mengi, M.; Aydın, M.S.; Eşrefoğlu, M.; et al. A comprehensive study of myocardial redox homeostasis in naturally and mimetically aged rats. Age 2014, 36, 9728.
- Chang, Y.M.; Chang, H.H.; Lin, H.J.; Tsai, C.C.; Tsai, C.T.; Chang, H.N.; Lin, S.L.; PadmaViswanadha, V.; Chen, R.J.; Huang, C.Y. Inhibition of Cardiac Hypertrophy Effects in D-Galactose-Induced Senescent Hearts by Alpinate Oxyphyllae Fructus Treatment. Evid. Based Complement Altern. Med. 2017, 2017, 2624384.
- Dehghani, A.; Hafizibarjin, Z.; Najjari, R.; Kaseb, F.; Safari, F. Resveratrol and 1,25-dihydroxyvitamin D co-administration protects the heart against D-galactose-induced aging in rats: Evaluation of serum and cardiac levels of klotho. Aging Clin. Exp. Res. 2019, 31, 1195–1205.
- Wang, L.F.; Cao, Q.; Wen, K.; Xiao, Y.F.; Chen, T.T.; Guan, X.H.; Liu, Y.; Zuo, L.; Qian, Y.S.; Deng, K.Y.; et al. CD38 Deficiency Alleviates D-Galactose-Induced Myocardial Cell Senescence Through NAD(+)/Sirt1 Signaling Pathway. Front. Physiol. 2019, 10, 1125.
- Lei, L.; Ou, L.; Yu, X. The antioxidant effect of Asparagus cochinchinensis (Lour.) Merr. shoot in D-galactose induced mice aging model and in vitro. J. Chin. Med. Assoc. 2016, 79, 205–211.
- Sun, S.L.; Guo, L.; Ren, Y.C.; Wang, B.; Li, R.H.; Qi, Y.S.; Yu, H.; Chang, N.D.; Li, M.H.; Peng, H.S. Anti-apoptosis effect of polysaccharide isolated from the seeds of Cuscuta chinensis Lam on cardiomyocytes in aging rats. Mol. Biol. Rep. 2014, 41, 6117–6124.
- Hegab, Z.; Gibbons, S.; Neyses, L.; Mamas, M.A. Role of advanced glycation end products in cardiovascular disease. World J. Cardiol. 2012, 4, 90–102.
- Bo-Htay, C.; Palee, S.; Apaijai, N.; Chattipakorn, S.C.; Chattipakorn, N. Effects of d-galactose-induced ageing on the heart and its potential interventions. J. Cell. Mol. Med. 2018, 22, 1392–1410.
- Chang, Y.M.; Tamilselvi, S.; Lin, H.J.; Tsai, C.C.; Lin, Y.M.; Day, C.H.; Viswanadha, V.P.; Chang, H.N.; Kuo, W.W.; Huang, C.Y. Alpinia oxyphylla Miq extract ameliorates cardiac fibrosis associated with D-galactose induced aging in rats. Environ. Toxicol. 2019, 34, 172–178.
- Yeh, S.L.; Wu, T.C.; Chan, S.T.; Hong, M.J.; Chen, H.L. Fructo-oligosaccharide attenuates the production of pro-inflammatory cytokines and the activation of JNK/Jun pathway in the lungs of D-galactose-treated Balb/cJ mice. Eur. J. Nutr. 2014, 53, 449–456.
- Feng, Y.; Yu, Y.H.; Wang, S.T.; Ren, J.; Camer, D.; Hua, Y.Z.; Zhang, Q.; Huang, J.; Xue, D.L.; Zhang, X.F.; et al. Chlorogenic acid protects D-galactose-induced liver and kidney injury via antioxidation and anti-inflammation effects in mice. Pharm. Biol. 2016, 54, 1027–1034.
- Chen, H.L.; Wang, C.H.; Kuo, Y.W.; Tsai, C.H. Antioxidative and hepatoprotective effects of fructo-oligosaccharide in d-galactose-treated Balb/cJ mice. Br. J. Nutr. 2011, 105, 805–809.
- Shahroudi, M.J.; Mehri, S.; Hosseinzadeh, H. Anti-Aging Effect of Nigella Sativa Fixed Oil on D-Galactose-Induced Aging in Mice. J. Pharmacopunct. 2017, 20, 29–35.
- Fan, Y.; Xia, J.; Jia, D.; Zhang, M.; Zhang, Y.; Huang, G.; Wang, Y. Mechanism of ginsenoside Rg1 renal protection in a mouse model of d-galactose-induced subacute damage. Pharm. Biol. 2016, 54, 1815–1821.
- Miller, M.R. Structural and physiological age-associated changes in aging lungs. Semin. Respir. Crit. Care Med. 2010, 31, 521–527.
- Gao, J.; Yu, Z.; Jing, S.; Jiang, W.; Liu, C.; Yu, C.; Sun, J.; Wang, C.; Chen, J.; Li, H. Protective effect of Anwulignan against D-galactose-induced hepatic injury through activating p38 MAPK-Nrf2-HO-1 pathway in mice. Clin. Interv. Aging 2018, 13, 1859–1869.
- Xu, L.Q.; Xie, Y.L.; Gui, S.H.; Zhang, X.; Mo, Z.Z.; Sun, C.Y.; Li, C.L.; Luo, D.D.; Zhang, Z.B.; Su, Z.R.; et al. Polydatin attenuates d-galactose-induced liver and brain damage through its anti-oxidative, anti-inflammatory and anti-apoptotic effects in mice. Food Funct. 2016, 7, 4545–4555.
- Zhang, X.; Wu, J.Z.; Lin, Z.X.; Yuan, Q.J.; Li, Y.C.; Liang, J.L.; Zhan, J.Y.; Xie, Y.L.; Su, Z.R.; Liu, Y.H. Ameliorative effect of supercritical fluid extract of Chrysanthemum indicum Linnén against D-galactose induced brain and liver injury in senescent mice via suppression of oxidative stress, inflammation and apoptosis. J. Ethnopharmacol. 2019, 234, 44–56.
- Huang, C.C.; Chiang, W.D.; Huang, W.C.; Huang, C.Y.; Hsu, M.C.; Lin, W.T. Hepatoprotective Effects of Swimming Exercise against D-Galactose-Induced Senescence Rat Model. Evid. Based Complement Altern. Med 2013, 2013, 275431.
- Wang, H.; Hu, L.; Li, L.; Wu, X.; Fan, Z.; Zhang, C.; Wang, J.; Jia, J.; Wang, S. Inorganic nitrate alleviates the senescence-related decline in liver function. Sci. China Life Sci. 2018, 61, 24–34.
- Mo, Z.Z.; Liu, Y.H.; Li, C.L.; Xu, L.Q.; Wen, L.L.; Xian, Y.F.; Lin, Z.X.; Zhan, J.Y.; Chen, J.N.; Xu, F.F.; et al. Protective Effect of SFE-CO2 of Ligusticum chuanxiong Hort Against d-Galactose-Induced Injury in the Mouse Liver and Kidney. Rejuvenation Res. 2017, 20, 231–243.
- Liu, C.M.; Ma, J.Q.; Lou, Y. Chronic administration of troxerutin protects mouse kidney against D-galactose-induced oxidative DNA damage. Food Chem. Toxicol. 2010, 48, 2809–2817.
- Zhuang, Y.; Ma, Q.; Guo, Y.; Sun, L. Protective effects of rambutan (Nephelium lappaceum) peel phenolics on H2O2-induced oxidative damages in HepG2 cells and d-galactose-induced aging mice. Food Chem. Toxicol. 2017, 108, 554–562.
- Liao, C.H.; Chen, B.H.; Chiang, H.S.; Chen, C.W.; Chen, M.F.; Ke, C.C.; Wang, Y.Y.; Lin, W.N.; Wang, C.C.; Lin, Y.H. Optimizing a Male Reproductive Aging Mouse Model by D-Galactose Injection. Int. J. Mol. Sci. 2016, 17, 98.
- Djahanbakhch, O.; Ezzati, M.; Zosmer, A. Reproductive ageing in women. J. Pathol. 2007, 211, 219–231.
- Ahangarpour, A.; Najimi, S.A.; Farbood, Y. Effects of Vitex agnus-castus fruit on sex hormones and antioxidant indices in a d-galactose-induced aging female mouse model. J. Chin. Med. Assoc. 2016, 79, 589–596.
- Wang, F.; Zhou, H.; Deng, L.; Wang, L.; Chen, J.; Zhou, X. Serine Deficiency Exacerbates Inflammation and Oxidative Stress via Microbiota-Gut-Brain Axis in D-Galactose-Induced Aging Mice. Mediat. Inflamm 2020, 2020, 5821428.
- Liu, X.; Wu, C.; Han, D.; Liu, J.; Liu, H.; Jiang, Z. Partially Hydrolyzed Guar Gum Attenuates d-Galactose-Induced Oxidative Stress and Restores Gut Microbiota in Rats. Int. J. Mol. Sci. 2019, 20, 4861.
- Fransen, F.; van Beek, A.A.; Borghuis, T.; Aidy, S.E.; Hugenholtz, F.; van der Gaast-de Jongh, C.; Savelkoul, H.F.J.; De Jonge, M.I.; Boekschoten, M.V.; Smidt, H.; et al. Aged Gut Microbiota Contributes to Systemical Inflammaging after Transfer to Germ-Free Mice. Front. Immunol. 2017, 8, 1385.
- Yin, R.; Liu, S.; Jiang, X.; Zhang, X.; Wei, F.; Hu, J. The Qingchangligan Formula Alleviates Acute Liver Failure by Regulating Galactose Metabolism and Gut Microbiota. Front. Cell Infect Microbiol. 2021, 11, 771483.
- Kim, S.; Jazwinski, S.M. The Gut Microbiota and Healthy Aging: A Mini-Review. Gerontology 2018, 64, 513–520.
- Takeda, T.; Hosokawa, M.; Higuchi, K. Senescence-accelerated mouse (SAM): A novel murine model of senescence. Exp. Gerontol. 1997, 32, 105–109.
- Higuchi, K. Genetic characterization of senescence-accelerated mouse (SAM). Exp. Gerontol. 1997, 32, 129–138.
- Garcia-Matas, S.; Gutierrez-Cuesta, J.; Coto-Montes, A.; Rubio-Acero, R.; Diez-Vives, C.; Camins, A.; Pallas, M.; Sanfeliu, C.; Cristofol, R. Dysfunction of astrocytes in senescence-accelerated mice SAMP8 reduces their neuroprotective capacity. Aging Cell 2008, 7, 630–640.
- Beck, J.; Horikawa, I.; Harris, C. Cellular Senescence: Mechanisms, Morphology, and Mouse Models. Vet. Pathol. 2020, 57, 747–757.
- Lecka-Czernik, B.; Moerman, E.J.; Shmookler Reis, R.J.; Lipschitz, D.A. Cellular and molecular biomarkers indicate precocious in vitro senescence in fibroblasts from SAMP6 mice. Evidence supporting a murine model of premature senescence and osteopenia. J. Gerontol. Biol. Sci. Med. Sci. 1997, 52, B331–B336.
- Manczak, M.; Jung, Y.; Park, B.S.; Partovi, D.; Reddy, P.H. Time-course of mitochondrial gene expressions in mice brains: Implications for mitochondrial dysfunction, oxidative damage, and cytochrome c in aging. J. Neurochem. 2005, 92, 494–504.
- Takeda, T.; Hosokawa, M.; Takeshita, S.; Irino, M.; Higuchi, K.; Matsushita, T.; Tomita, Y.; Yasuhira, K.; Hamamoto, H.; Shimizu, K.; et al. A new murine model of accelerated senescence. Mech. Ageing Dev. 1981, 17, 183–194.
- Takeda, T.; Hosokawa, M.; Higuchi, K. Senescence-accelerated mouse (SAM): A novel murine model of accelerated senescence. J. Am. Geriatr. Soc. 1991, 39, 911–919.
- Matsushita, M.; Tsuboyama, T.; Kasai, R.; Okumura, H.; Yamamuro, T.; Higuchi, K.; Higuchi, K.; Kohno, A.; Yonezu, T.; Utani, A.; et al. Age-related changes in bone mass in the senescence-accelerated mouse (SAM). SAM-R/3 and SAM-P/6 as new murine models for senile osteoporosis. Am. J. Pathol. 1986, 125, 276–283.
- Takada, K.; Inaba, M.; Ichioka, N.; Ueda, Y.; Taira, M.; Baba, S.; Mizokami, T.; Wang, X.; Hisha, H.; Iida, H.; et al. Treatment of senile osteoporosis in SAMP6 mice by intra-bone marrow injection of allogeneic bone marrow cells. Stem Cells 2006, 24, 399–405.
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulić, V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117.
- Al-Azab, M.; Wang, B.; Elkhider, A.; Walana, W.; Li, W.; Yuan, B.; Ye, Y.; Tang, Y.; Almoiliqy, M.; Adlat, S.; et al. Indian Hedgehog regulates senescence in bone marrow-derived mesenchymal stem cell through modulation of ROS/mTOR/4EBP1, p70S6K1/2 pathway. Aging 2020, 12, 5693–5715.
- Morley, J.E.; Armbrecht, H.J.; Farr, S.A.; Kumar, V.B. The senescence accelerated mouse (SAMP8) as a model for oxidative stress and Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 650–656.
- Karasawa, N.; Nagatsu, I.; Sakai, K.; Nagatsu, T.; Watanabe, K.; Onozuka, M. Immunocytochemical study of catecholaminergic neurons in the senescence-accelerated mouse (SAM-P8) brain. J. Neural Transm. 1997, 104, 1267–1275.
- Sureda, F.X.; Gutierrez-Cuesta, J.; Romeu, M.; Mulero, M.; Canudas, A.M.; Camins, A.; Mallol, J.; Pallas, M. Changes in oxidative stress parameters and neurodegeneration markers in the brain of the senescence-accelerated mice SAMP-8. Exp. Gerontol. 2006, 41, 360–367.
- Manich, G.; Mercader, C.; del Valle, J.; Duran-Vilaregut, J.; Camins, A.; Pallàs, M.; Vilaplana, J.; Pelegrí, C. Characterization of amyloid-β granules in the hippocampus of SAMP8 mice. J. Alzheimers Dis. 2011, 25, 535–546.
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38, e100492.
- Alvarez-Garcia, O.; Vega-Naredo, I.; Sierra, V.; Caballero, B.; Tomas-Zapico, C.; Camins, A.; Garcia, J.J.; Pallas, M.; Coto-Montes, A. Elevated oxidative stress in the brain of senescence-accelerated mice at 5 months of age. Biogerontology 2006, 7, 43–52.
- Ota, H.; Akishita, M.; Akiyoshi, T.; Kahyo, T.; Setou, M.; Ogawa, S.; Iijima, K.; Eto, M.; Ouchi, Y. Testosterone deficiency accelerates neuronal and vascular aging of SAMP8 mice: Protective role of eNOS and SIRT1. PLoS ONE 2012, 7, e29598.
- Karuppagounder, V.; Arumugam, S.; Babu, S.S.; Palaniyandi, S.S.; Watanabe, K.; Cooke, J.P.; Thandavarayan, R.A. The senescence accelerated mouse prone 8 (SAMP8): A novel murine model for cardiac aging. Ageing Res. Rev. 2017, 35, 291–296.
- Niimi, K.; Takahashi, E.; Itakura, C. Adiposity-related biochemical phenotype in senescence-accelerated mouse prone 6 (SAMP6). Comp. Med. 2009, 59, 431–436.
- Shimada, A.; Ohta, A.; Akiguchi, I.; Takeda, T. Inbred SAM-P/10 as a mouse model of spontaneous, inherited brain atrophy. J. Neuropathol. Exp. Neurol. 1992, 51, 440–450.
- Shimada, A.; Hosokawa, M.; Ohta, A.; Akiguchi, I.; Takeda, T. Localization of atrophy-prone areas in the aging mouse brain: Comparison between the brain atrophy model SAM-P/10 and the normal control SAM-R/1. Neuroscience 1994, 59, 859–869.
- Shcherbakov, D.; Nigri, M.; Akbergenov, R.; Brilkova, M.; Mantovani, M.; Petit, P.I.; Grimm, A.; Karol, A.A.; Teo, Y.; Sanchón, A.C.; et al. Premature aging in mice with error-prone protein synthesis. Sci. Adv. 2022, 8, eabl9051.
- Brennan, T.A.; Egan, K.P.; Lindborg, C.M.; Chen, Q.; Sweetwyne, M.T.; Hankenson, K.D.; Xie, S.X.; Johnson, F.B.; Pignolo, R.J. Mouse models of telomere dysfunction phenocopy skeletal changes found in human age-related osteoporosis. Dis. Model Mech. 2014, 7, 583–592.
- Botter, S.M.; Zar, M.; van Osch, G.J.; van Steeg, H.; Dolle, M.E.; Hoeijmakers, J.H.; Weinans, H.; van Leeuwen, J.P. Analysis of osteoarthritis in a mouse model of the progeroid human DNA repair syndrome trichothiodystrophy. Age 2011, 33, 247–260.
- Edstrom, E.; Altun, M.; Bergman, E.; Johnson, H.; Kullberg, S.; Ramirez-Leon, V.; Ulfhake, B. Factors contributing to neuromuscular impairment and sarcopenia during aging. Physiol. Behav. 2007, 92, 129–135.
- Baker, D.J.; Jeganathan, K.B.; Cameron, J.D.; Thompson, M.; Juneja, S.; Kopecka, A.; Kumar, R.; Jenkins, R.B.; de Groen, P.C.; Roche, P.; et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 2004, 36, 744–749.
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189.
- Hartman, T.K.; Wengenack, T.M.; Poduslo, J.F.; van Deursen, J.M. Mutant mice with small amounts of BubR1 display accelerated age-related gliosis. Neurobiol. Aging 2007, 28, 921–927.
- Loerch, P.M.; Lu, T.; Dakin, K.A.; Vann, J.M.; Isaacs, A.; Geula, C.; Wang, J.; Pan, Y.; Gabuzda, D.H.; Li, C.; et al. Evolution of the aging brain transcriptome and synaptic regulation. PLoS ONE 2008, 3, e3329.
- Bishop, N.A.; Lu, T.; Yankner, B.A. Neural mechanisms of ageing and cognitive decline. Nature 2010, 464, 529–535.
- Esquerda-Canals, G.; Montoliu-Gaya, L.; Güell-Bosch, J.; Villegas, S. Mouse Models of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1171–1183.
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular Basis of Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 952–963.
- Teich, A.F.; Patel, M.; Arancio, O. A reliable way to detect endogenous murine β-amyloid. PLoS ONE 2013, 8, e55647.
- Myers, A.; McGonigle, P. Overview of Transgenic Mouse Models for Alzheimer’s Disease. Curr. Protoc. Neurosci. 2019, 89, e81.
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Berthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 1995, 373, 523–527.
- Hall, A.M.; Roberson, E.D. Mouse models of Alzheimer’s disease. Brain Res. Bull 2012, 88, 3–12.
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487.
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421.
- Zhang, X.; Luhrs, K.J.; Ryff, K.A.; Malik, W.T.; Driscoll, M.J.; Culver, B. Suppression of nuclear factor kappa B ameliorates astrogliosis but not amyloid burden in APPswe/PS1dE9 mice. Neuroscience 2009, 161, 53–58.
- Sun, X.-Y.; Li, L.-J.; Dong, Q.-X.; Zhu, J.; Huang, Y.-R.; Hou, S.-J.; Yu, X.-L.; Liu, R.-T. Rutin prevents tau pathology and neuroinflammation in a mouse model of Alzheimer’s disease. J. Neuroinflamm. 2021, 18, 131.
- Xie, W.-Q.; He, M.; Yu, D.-J.; Wu, Y.-X.; Wang, X.-H.; Lv, S.; Xiao, W.-F.; Li, Y.-S. Mouse models of sarcopenia: Classification and evaluation. J. Cachexia Sarcopenia Muscle 2021, 12, 538–554.
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693.
- Capitanio, D.; Moriggi, M.; De Palma, S.; Bizzotto, D.; Molon, S.; Torretta, E.; Fania, C.; Bonaldo, P.; Gelfi, C.; Braghetta, P. Collagen VI Null Mice as a Model for Early Onset Muscle Decline in Aging. Front. Mol. Neurosci. 2017, 10, 337.
- Ko, F.; Abadir, P.; Marx, R.; Westbrook, R.; Cooke, C.; Yang, H.; Walston, J. Impaired mitochondrial degradation by autophagy in the skeletal muscle of the aged female interleukin 10 null mouse. Exp. Gerontol. 2016, 73, 23–27.
- Ahn, B.; Smith, N.; Saunders, D.; Ranjit, R.; Kneis, P.; Towner, R.A.; Van Remmen, H. Using MRI to measure in vivo free radical production and perfusion dynamics in a mouse model of elevated oxidative stress and neurogenic atrophy. Redox. Biol. 2019, 26, 101308.
- Sayed, R.K.A.; Fernández-Ortiz, M.; Diaz-Casado, M.E.; Aranda-Martínez, P.; Fernández-Martínez, J.; Guerra-Librero, A.; Escames, G.; López, L.C.; Alsaadawy, R.M.; Acuña-Castroviejo, D. Lack of NLRP3 Inflammasome Activation Reduces Age-Dependent Sarcopenia and Mitochondrial Dysfunction, Favoring the Prophylactic Effect of Melatonin. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2019, 74, 1699–1708.
- Li, J.; Yi, X.; Yao, Z.; Chakkalakal, J.V.; Xing, L.; Boyce, B.F. TNF Receptor-Associated Factor 6 Mediates TNFα-Induced Skeletal Muscle Atrophy in Mice During Aging. J. Bone Min. Res. 2020, 35, 1535–1548.
- Sun, H.; Gong, Y.; Qiu, J.; Chen, Y.; Ding, F.; Zhao, Q. TRAF6 inhibition rescues dexamethasone-induced muscle atrophy. Int. J. Mol. Sci. 2014, 15, 11126–11141.
- Wada, S.; Kato, Y.; Okutsu, M.; Miyaki, S.; Suzuki, K.; Yan, Z.; Schiaffino, S.; Asahara, H.; Ushida, T.; Akimoto, T. Translational suppression of atrophic regulators by microRNA-23a integrates resistance to skeletal muscle atrophy. J. Biol. Chem. 2011, 286, 38456–38465.
- Campos, F.; Abrigo, J.; Aguirre, F.; Garcés, B.; Arrese, M.; Karpen, S.; Cabrera, D.; Andía, M.E.; Simon, F.; Cabello-Verrugio, C. Sarcopenia in a mice model of chronic liver disease: Role of the ubiquitin-proteasome system and oxidative stress. Pflug. Arch. Eur. J. Physiol. 2018, 470, 1503–1519.
- Noll, N.A.; Lal, H.; Merryman, W.D. Mouse Models of Heart Failure with Preserved or Reduced Ejection Fraction. Am. J. Pathol. 2020, 190, 1596–1608.
- Meng, Q.; Lai, Y.C.; Kelly, N.J.; Bueno, M.; Baust, J.J.; Bachman, T.N.; Goncharov, D.; Vanderpool, R.R.; Radder, J.E.; Hu, J.; et al. Development of a Mouse Model of Metabolic Syndrome, Pulmonary Hypertension, and Heart Failure with Preserved Ejection Fraction. Am. J. Respir. Cell Mol. Biol. 2017, 56, 497–505.
- Manrique, C.; DeMarco, V.G.; Aroor, A.R.; Mugerfeld, I.; Garro, M.; Habibi, J.; Hayden, M.R.; Sowers, J.R. Obesity and insulin resistance induce early development of diastolic dysfunction in young female mice fed a Western diet. Endocrinology 2013, 154, 3632–3642.
- Aroor, A.R.; Habibi, J.; Kandikattu, H.K.; Garro-Kacher, M.; Barron, B.; Chen, D.; Hayden, M.R.; Whaley-Connell, A.; Bender, S.B.; Klein, T.; et al. Dipeptidyl peptidase-4 (DPP-4) inhibition with linagliptin reduces western diet-induced myocardial TRAF3IP2 expression, inflammation and fibrosis in female mice. Cardiovasc. Diabetol. 2017, 16, 61.
- Cannon, M.V.; Silljé, H.H.; Sijbesma, J.W.; Khan, M.A.; Steffensen, K.R.; van Gilst, W.H.; de Boer, R.A. LXRα improves myocardial glucose tolerance and reduces cardiac hypertrophy in a mouse model of obesity-induced type 2 diabetes. Diabetologia 2016, 59, 634–643.
- Withaar, C.; Meems, L.M.G.; Markousis-Mavrogenis, G.; Boogerd, C.J.; Silljé, H.H.W.; Schouten, E.M.; Dokter, M.M.; Voors, A.A.; Westenbrink, B.D.; Lam, C.S.P.; et al. The effects of liraglutide and dapagliflozin on cardiac function and structure in a multi-hit mouse model of heart failure with preserved ejection fraction. Cardiovasc. Res. 2021, 117, 2108–2124.
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.1K
Revisions:
2 times
(View History)
Update Date:
05 May 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No