With the global increase of the elderly population, the improvement of the treatment for various aging-related diseases and the extension of a healthy lifespan have become some of the most important current medical issues. In order to understand the developmental mechanisms of aging and aging-related disorders, animal models are essential to conduct relevant studies. Among them, mice have become one of the most prevalently used model animals for aging-related studies due to their high similarity to humans in terms of genetic background and physiological structure, as well as their short lifespan and ease of reproduction.
- aging
- aging-related disease
- mouse model
1. Introduction
2. Systemic-Induced Accelerated Aging Mouse Model
| Type | Subdivision | Phenotypes |
|---|---|---|
| D-galactose-induced senescence model | Brain | Cognitive impairment Mitochondrial dysfunction Neuronal degeneration Apoptosis Depressive and anxious |
| Heart | Cardiac fibrosis Collagen accumulation Fibroblasts disordered arrangement |
|
| Kidney | Kidney index ↓ Uric acid & Cys-C ↑ Glomerular and tubular damage ↑ |
|
| Liver | Liver fibrosis Glycogen levels ↓ Lipid deposition ↑ |
|
| Reproductive system |
Estrogen and progesterone ↓ Ovarian follicle regression Uterine wall endometrial gland atrophy Disrupt estrous cycles |
|
| Intestinal flora | Disturbance | |
| Lung | Oxidative stress ↑ Fibrotic status Chronic inflammation |
|
| SAMP mice | SAMP 1 | Aging amyloidosis Immune dysfunction Renal atrophy Hearing loss Senile pulmonary hyperinflation |
| SAMP 6 | Senile osteoporosis Myeloid progenitor cell senescence |
|
| SAMP 8 | Astrogliosis Microgliosis Neurodegeneration Amyloid accumulation MAPT hyperphosphorylation |
|
| SAMP 10 | Learning and memory impairment Cerebral cortex and limbic system atrophy |
|
| Rps9 D95N mouse | Altered fur Cataracts Hunched posture Body composition function & body weight ↓ Fat mass & muscle strength ↓ Shortened lifespan Mouse urinary syndrome Extramedullary hematopoiesis |
|
| Lama−/− | Short lifespan Growth retardation Muscular dystrophy Altered lipid metabolism |
|
| Wrn∆hel/∆hel | Short lifespan Abnormal hyaluronic acid excretion Metabolic abnormalities Increased genomic instability and cancer incidence |
|
| Csa−/−, Csb−/− | Short lifespan Reduced fat mass Photoreceptor cell loss Neural pathology |
|
| Progeria syndrome mouse | XpdTTD/TTD | Short lifespan Trichothiodystrophy |
| Bub1bH/H, Xpg−/− | Brain atrophy Neuronal loss Neurofibrillary deposition of Aβ or senile plaques |
|
| Bub1bH/H, Bub1b+/GTTA | Mean muscle fiber diameter ↓ Muscle fiber size variation ↑ Intermuscular fibrosis Regenerative capacity of skeletal muscles ↓ |
|
| Mitochondrial DNA polymerase mutant mouse | Lifespan ↓ Weight loss Subcutaneous fat ↓ Hair loss Kyphosis Osteoporosis Anemia Fertility ↓ Spermatogonia depletion Heart enlargement |
|
| Total body irradiation (TBI) model | Progressive premature frailty Cognitive decline Whole blood antioxidant capacity ↓ RBC glutathione ↓ Thymic involution Articular cartilage and bone degeneration Ovarian environment damage |
|
| Ozone-induced senescence model | Cognitive decline Memory impairment AD symptoms Lung tumor growth ↑ |
|
| Chronic jet-lag mouse | Accelerated initial tumor growth Shortened mouse survival Induce spontaneous hepatocellular carcinoma Obesity Depression Addiction Abnormal cardiac structure Impaired cardiac function |
2.1. The D-Galactose-Induced Senescence Model
2.2. Senescence-Accelerated Mouse/Prone
2.3. Rps9 D95N Mouse
2.4. Progeria Syndrome Mouse
3. Tissue-, Organ-, or System-Specific Mouse Models of Aging-Related Diseases
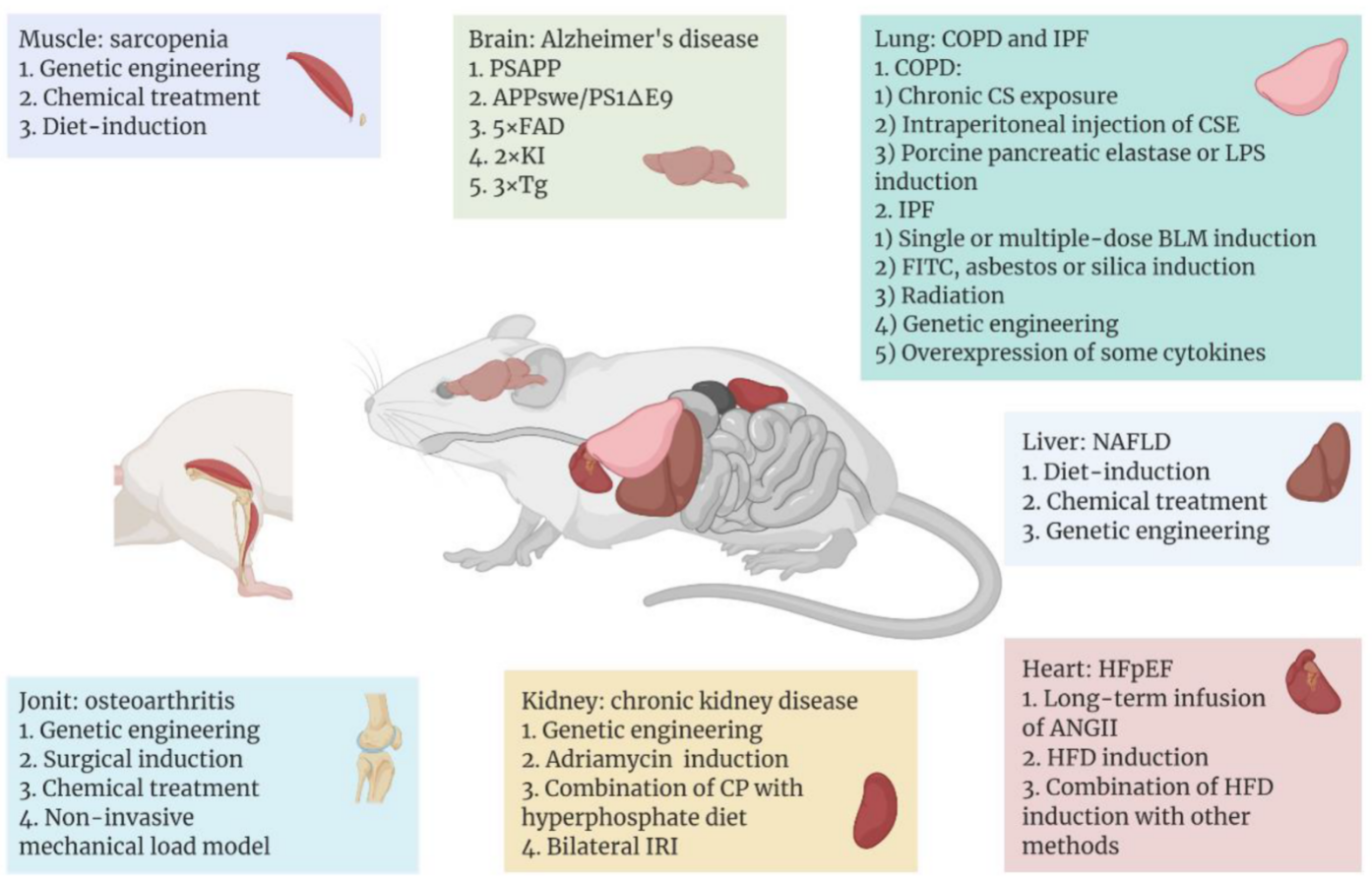
3.1. Model of Aging Brain or Nerve System
3.2. Model of Aging Muscle
3.3. Model of Aging Heart
This entry is adapted from the peer-reviewed paper 10.3390/cells11091418
References
- DESA|United Nations. Available online: https://www.un.org/en/desa (accessed on 20 March 2022).
- World Population Prospects 2019. Available online: https://population.un.org/wpp/ (accessed on 20 March 2022).
- Graw, J. Mouse models of cataract. J. Genet. 2009, 88, 469–486.
- Brooks, S.V.; Faulkner, J.A. Skeletal muscle weakness in old age: Underlying mechanisms. Med. Sci. Sports Exerc. 1994, 26, 432–439.
- Gurkar, A.U.; Niedernhofer, L.J. Comparison of mice with accelerated aging caused by distinct mechanisms. Exp. Gerontol. 2015, 68, 43–50.
- Vanhooren, V.; Libert, C. The mouse as a model organism in aging research: Usefulness, pitfalls and possibilities. Ageing Res. Rev. 2013, 12, 8–21.
- Harkema, L.; Youssef, S.A.; de Bruin, A. Pathology of Mouse Models of Accelerated Aging. Vet. Pathol. 2016, 53, 366–389.
- Acosta, P.B.; Gross, K.C. Hidden sources of galactose in the environment. Eur. J. Pediatrics 1995, 154, S87–S92.
- Morava, E. Galactose supplementation in phosphoglucomutase-1 deficiency; review and outlook for a novel treatable CDG. Mol Genet. Metab. 2014, 112, 275–279.
- Ullah, F.; Ali, T.; Ullah, N.; Kim, M.O. Caffeine prevents d-galactose-induced cognitive deficits, oxidative stress, neuroinflammation and neurodegeneration in the adult rat brain. Neurochem. Int. 2015, 90, 114–124.
- Wu, D.M.; Lu, J.; Zheng, Y.L.; Zhou, Z.; Shan, Q.; Ma, D.F. Purple sweet potato color repairs d-galactose-induced spatial learning and memory impairment by regulating the expression of synaptic proteins. Neurobiol. Learn Mem. 2008, 90, 19–27.
- Azman, K.F.; Safdar, A.; Zakaria, R. D-galactose-induced liver aging model: Its underlying mechanisms and potential therapeutic interventions. Exp. Gerontol. 2021, 150, 111372.
- Shwe, T.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. Role of D-galactose-induced brain aging and its potential used for therapeutic interventions. Exp. Gerontol. 2018, 101, 13–36.
- Azman, K.F.; Zakaria, R. D-Galactose-induced accelerated aging model: An overview. Biogerontology 2019, 20, 763–782.
- Li, W.; Wang, S.; Wang, H.; Wang, J.; Jin, F.; Fang, F.; Fang, C. Astragaloside IV Prevents Memory Impairment in D-galactose-induced Aging Rats Via the AGEs/RAGE/ NF-kappaB Axis. Arch. Med. Res. 2022, 53, 20–28.
- Banji, O.J.; Banji, D.; Ch, K. Curcumin and hesperidin improve cognition by suppressing mitochondrial dysfunction and apoptosis induced by D-galactose in rat brain. Food Chem. Toxicol. 2014, 74, 51–59.
- Khedr, N.F.; Werida, R.H.; Abo-Saif, M.A. Candesartan protects against d-galactose induced—Neurotoxicity and memory deficit via modulation of autophagy and oxidative stress. Toxicol. Appl. Pharm. 2022, 435, 115827.
- Long, J.; Wang, X.; Gao, H.; Liu, Z.; Liu, C.; Miao, M.; Cui, X.; Packer, L.; Liu, J. D-galactose toxicity in mice is associated with mitochondrial dysfunction: Protecting effects of mitochondrial nutrient R-alpha-lipoic acid. Biogerontology 2007, 8, 373–381.
- Kumar, A.; Dogra, S.; Prakash, A. Effect of carvedilol on behavioral, mitochondrial dysfunction, and oxidative damage against D-galactose induced senescence in mice. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2009, 380, 431–441.
- Haider, S.; Liaquat, L.; Shahzad, S.; Sadir, S.; Madiha, S.; Batool, Z.; Tabassum, S.; Saleem, S.; Naqvi, F.; Perveen, T. A high dose of short term exogenous D-galactose administration in young male rats produces symptoms simulating the natural aging process. Life Sci. 2015, 124, 110–119.
- Sacco, R.L.; Roth, G.A.; Reddy, K.S.; Arnett, D.K.; Bonita, R.; Gaziano, T.A.; Heidenreich, P.A.; Huffman, M.D.; Mayosi, B.M.; Mendis, S.; et al. The Heart of 25 by 25: Achieving the Goal of Reducing Global and Regional Premature Deaths from Cardiovascular Diseases and Stroke: A Modeling Study from the American Heart Association and World Heart Federation. Circulation 2016, 133, e674–e690.
- Cebe, T.; Yanar, K.; Atukeren, P.; Ozan, T.; Kuruç, A.I.; Kunbaz, A.; Sitar, M.E.; Mengi, M.; Aydın, M.S.; Eşrefoğlu, M.; et al. A comprehensive study of myocardial redox homeostasis in naturally and mimetically aged rats. Age 2014, 36, 9728.
- Chang, Y.M.; Chang, H.H.; Lin, H.J.; Tsai, C.C.; Tsai, C.T.; Chang, H.N.; Lin, S.L.; PadmaViswanadha, V.; Chen, R.J.; Huang, C.Y. Inhibition of Cardiac Hypertrophy Effects in D-Galactose-Induced Senescent Hearts by Alpinate Oxyphyllae Fructus Treatment. Evid. Based Complement Altern. Med. 2017, 2017, 2624384.
- Dehghani, A.; Hafizibarjin, Z.; Najjari, R.; Kaseb, F.; Safari, F. Resveratrol and 1,25-dihydroxyvitamin D co-administration protects the heart against D-galactose-induced aging in rats: Evaluation of serum and cardiac levels of klotho. Aging Clin. Exp. Res. 2019, 31, 1195–1205.
- Wang, L.F.; Cao, Q.; Wen, K.; Xiao, Y.F.; Chen, T.T.; Guan, X.H.; Liu, Y.; Zuo, L.; Qian, Y.S.; Deng, K.Y.; et al. CD38 Deficiency Alleviates D-Galactose-Induced Myocardial Cell Senescence Through NAD(+)/Sirt1 Signaling Pathway. Front. Physiol. 2019, 10, 1125.
- Lei, L.; Ou, L.; Yu, X. The antioxidant effect of Asparagus cochinchinensis (Lour.) Merr. shoot in D-galactose induced mice aging model and in vitro. J. Chin. Med. Assoc. 2016, 79, 205–211.
- Sun, S.L.; Guo, L.; Ren, Y.C.; Wang, B.; Li, R.H.; Qi, Y.S.; Yu, H.; Chang, N.D.; Li, M.H.; Peng, H.S. Anti-apoptosis effect of polysaccharide isolated from the seeds of Cuscuta chinensis Lam on cardiomyocytes in aging rats. Mol. Biol. Rep. 2014, 41, 6117–6124.
- Hegab, Z.; Gibbons, S.; Neyses, L.; Mamas, M.A. Role of advanced glycation end products in cardiovascular disease. World J. Cardiol. 2012, 4, 90–102.
- Bo-Htay, C.; Palee, S.; Apaijai, N.; Chattipakorn, S.C.; Chattipakorn, N. Effects of d-galactose-induced ageing on the heart and its potential interventions. J. Cell. Mol. Med. 2018, 22, 1392–1410.
- Chang, Y.M.; Tamilselvi, S.; Lin, H.J.; Tsai, C.C.; Lin, Y.M.; Day, C.H.; Viswanadha, V.P.; Chang, H.N.; Kuo, W.W.; Huang, C.Y. Alpinia oxyphylla Miq extract ameliorates cardiac fibrosis associated with D-galactose induced aging in rats. Environ. Toxicol. 2019, 34, 172–178.
- Yeh, S.L.; Wu, T.C.; Chan, S.T.; Hong, M.J.; Chen, H.L. Fructo-oligosaccharide attenuates the production of pro-inflammatory cytokines and the activation of JNK/Jun pathway in the lungs of D-galactose-treated Balb/cJ mice. Eur. J. Nutr. 2014, 53, 449–456.
- Feng, Y.; Yu, Y.H.; Wang, S.T.; Ren, J.; Camer, D.; Hua, Y.Z.; Zhang, Q.; Huang, J.; Xue, D.L.; Zhang, X.F.; et al. Chlorogenic acid protects D-galactose-induced liver and kidney injury via antioxidation and anti-inflammation effects in mice. Pharm. Biol. 2016, 54, 1027–1034.
- Chen, H.L.; Wang, C.H.; Kuo, Y.W.; Tsai, C.H. Antioxidative and hepatoprotective effects of fructo-oligosaccharide in d-galactose-treated Balb/cJ mice. Br. J. Nutr. 2011, 105, 805–809.
- Shahroudi, M.J.; Mehri, S.; Hosseinzadeh, H. Anti-Aging Effect of Nigella Sativa Fixed Oil on D-Galactose-Induced Aging in Mice. J. Pharmacopunct. 2017, 20, 29–35.
- Fan, Y.; Xia, J.; Jia, D.; Zhang, M.; Zhang, Y.; Huang, G.; Wang, Y. Mechanism of ginsenoside Rg1 renal protection in a mouse model of d-galactose-induced subacute damage. Pharm. Biol. 2016, 54, 1815–1821.
- Miller, M.R. Structural and physiological age-associated changes in aging lungs. Semin. Respir. Crit. Care Med. 2010, 31, 521–527.
- Gao, J.; Yu, Z.; Jing, S.; Jiang, W.; Liu, C.; Yu, C.; Sun, J.; Wang, C.; Chen, J.; Li, H. Protective effect of Anwulignan against D-galactose-induced hepatic injury through activating p38 MAPK-Nrf2-HO-1 pathway in mice. Clin. Interv. Aging 2018, 13, 1859–1869.
- Xu, L.Q.; Xie, Y.L.; Gui, S.H.; Zhang, X.; Mo, Z.Z.; Sun, C.Y.; Li, C.L.; Luo, D.D.; Zhang, Z.B.; Su, Z.R.; et al. Polydatin attenuates d-galactose-induced liver and brain damage through its anti-oxidative, anti-inflammatory and anti-apoptotic effects in mice. Food Funct. 2016, 7, 4545–4555.
- Zhang, X.; Wu, J.Z.; Lin, Z.X.; Yuan, Q.J.; Li, Y.C.; Liang, J.L.; Zhan, J.Y.; Xie, Y.L.; Su, Z.R.; Liu, Y.H. Ameliorative effect of supercritical fluid extract of Chrysanthemum indicum Linnén against D-galactose induced brain and liver injury in senescent mice via suppression of oxidative stress, inflammation and apoptosis. J. Ethnopharmacol. 2019, 234, 44–56.
- Huang, C.C.; Chiang, W.D.; Huang, W.C.; Huang, C.Y.; Hsu, M.C.; Lin, W.T. Hepatoprotective Effects of Swimming Exercise against D-Galactose-Induced Senescence Rat Model. Evid. Based Complement Altern. Med 2013, 2013, 275431.
- Wang, H.; Hu, L.; Li, L.; Wu, X.; Fan, Z.; Zhang, C.; Wang, J.; Jia, J.; Wang, S. Inorganic nitrate alleviates the senescence-related decline in liver function. Sci. China Life Sci. 2018, 61, 24–34.
- Mo, Z.Z.; Liu, Y.H.; Li, C.L.; Xu, L.Q.; Wen, L.L.; Xian, Y.F.; Lin, Z.X.; Zhan, J.Y.; Chen, J.N.; Xu, F.F.; et al. Protective Effect of SFE-CO2 of Ligusticum chuanxiong Hort Against d-Galactose-Induced Injury in the Mouse Liver and Kidney. Rejuvenation Res. 2017, 20, 231–243.
- Liu, C.M.; Ma, J.Q.; Lou, Y. Chronic administration of troxerutin protects mouse kidney against D-galactose-induced oxidative DNA damage. Food Chem. Toxicol. 2010, 48, 2809–2817.
- Zhuang, Y.; Ma, Q.; Guo, Y.; Sun, L. Protective effects of rambutan (Nephelium lappaceum) peel phenolics on H2O2-induced oxidative damages in HepG2 cells and d-galactose-induced aging mice. Food Chem. Toxicol. 2017, 108, 554–562.
- Liao, C.H.; Chen, B.H.; Chiang, H.S.; Chen, C.W.; Chen, M.F.; Ke, C.C.; Wang, Y.Y.; Lin, W.N.; Wang, C.C.; Lin, Y.H. Optimizing a Male Reproductive Aging Mouse Model by D-Galactose Injection. Int. J. Mol. Sci. 2016, 17, 98.
- Djahanbakhch, O.; Ezzati, M.; Zosmer, A. Reproductive ageing in women. J. Pathol. 2007, 211, 219–231.
- Ahangarpour, A.; Najimi, S.A.; Farbood, Y. Effects of Vitex agnus-castus fruit on sex hormones and antioxidant indices in a d-galactose-induced aging female mouse model. J. Chin. Med. Assoc. 2016, 79, 589–596.
- Wang, F.; Zhou, H.; Deng, L.; Wang, L.; Chen, J.; Zhou, X. Serine Deficiency Exacerbates Inflammation and Oxidative Stress via Microbiota-Gut-Brain Axis in D-Galactose-Induced Aging Mice. Mediat. Inflamm 2020, 2020, 5821428.
- Liu, X.; Wu, C.; Han, D.; Liu, J.; Liu, H.; Jiang, Z. Partially Hydrolyzed Guar Gum Attenuates d-Galactose-Induced Oxidative Stress and Restores Gut Microbiota in Rats. Int. J. Mol. Sci. 2019, 20, 4861.
- Fransen, F.; van Beek, A.A.; Borghuis, T.; Aidy, S.E.; Hugenholtz, F.; van der Gaast-de Jongh, C.; Savelkoul, H.F.J.; De Jonge, M.I.; Boekschoten, M.V.; Smidt, H.; et al. Aged Gut Microbiota Contributes to Systemical Inflammaging after Transfer to Germ-Free Mice. Front. Immunol. 2017, 8, 1385.
- Yin, R.; Liu, S.; Jiang, X.; Zhang, X.; Wei, F.; Hu, J. The Qingchangligan Formula Alleviates Acute Liver Failure by Regulating Galactose Metabolism and Gut Microbiota. Front. Cell Infect Microbiol. 2021, 11, 771483.
- Kim, S.; Jazwinski, S.M. The Gut Microbiota and Healthy Aging: A Mini-Review. Gerontology 2018, 64, 513–520.
- Takeda, T.; Hosokawa, M.; Higuchi, K. Senescence-accelerated mouse (SAM): A novel murine model of senescence. Exp. Gerontol. 1997, 32, 105–109.
- Higuchi, K. Genetic characterization of senescence-accelerated mouse (SAM). Exp. Gerontol. 1997, 32, 129–138.
- Garcia-Matas, S.; Gutierrez-Cuesta, J.; Coto-Montes, A.; Rubio-Acero, R.; Diez-Vives, C.; Camins, A.; Pallas, M.; Sanfeliu, C.; Cristofol, R. Dysfunction of astrocytes in senescence-accelerated mice SAMP8 reduces their neuroprotective capacity. Aging Cell 2008, 7, 630–640.
- Beck, J.; Horikawa, I.; Harris, C. Cellular Senescence: Mechanisms, Morphology, and Mouse Models. Vet. Pathol. 2020, 57, 747–757.
- Lecka-Czernik, B.; Moerman, E.J.; Shmookler Reis, R.J.; Lipschitz, D.A. Cellular and molecular biomarkers indicate precocious in vitro senescence in fibroblasts from SAMP6 mice. Evidence supporting a murine model of premature senescence and osteopenia. J. Gerontol. Biol. Sci. Med. Sci. 1997, 52, B331–B336.
- Manczak, M.; Jung, Y.; Park, B.S.; Partovi, D.; Reddy, P.H. Time-course of mitochondrial gene expressions in mice brains: Implications for mitochondrial dysfunction, oxidative damage, and cytochrome c in aging. J. Neurochem. 2005, 92, 494–504.
- Takeda, T.; Hosokawa, M.; Takeshita, S.; Irino, M.; Higuchi, K.; Matsushita, T.; Tomita, Y.; Yasuhira, K.; Hamamoto, H.; Shimizu, K.; et al. A new murine model of accelerated senescence. Mech. Ageing Dev. 1981, 17, 183–194.
- Takeda, T.; Hosokawa, M.; Higuchi, K. Senescence-accelerated mouse (SAM): A novel murine model of accelerated senescence. J. Am. Geriatr. Soc. 1991, 39, 911–919.
- Matsushita, M.; Tsuboyama, T.; Kasai, R.; Okumura, H.; Yamamuro, T.; Higuchi, K.; Higuchi, K.; Kohno, A.; Yonezu, T.; Utani, A.; et al. Age-related changes in bone mass in the senescence-accelerated mouse (SAM). SAM-R/3 and SAM-P/6 as new murine models for senile osteoporosis. Am. J. Pathol. 1986, 125, 276–283.
- Takada, K.; Inaba, M.; Ichioka, N.; Ueda, Y.; Taira, M.; Baba, S.; Mizokami, T.; Wang, X.; Hisha, H.; Iida, H.; et al. Treatment of senile osteoporosis in SAMP6 mice by intra-bone marrow injection of allogeneic bone marrow cells. Stem Cells 2006, 24, 399–405.
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulić, V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117.
- Al-Azab, M.; Wang, B.; Elkhider, A.; Walana, W.; Li, W.; Yuan, B.; Ye, Y.; Tang, Y.; Almoiliqy, M.; Adlat, S.; et al. Indian Hedgehog regulates senescence in bone marrow-derived mesenchymal stem cell through modulation of ROS/mTOR/4EBP1, p70S6K1/2 pathway. Aging 2020, 12, 5693–5715.
- Morley, J.E.; Armbrecht, H.J.; Farr, S.A.; Kumar, V.B. The senescence accelerated mouse (SAMP8) as a model for oxidative stress and Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 650–656.
- Karasawa, N.; Nagatsu, I.; Sakai, K.; Nagatsu, T.; Watanabe, K.; Onozuka, M. Immunocytochemical study of catecholaminergic neurons in the senescence-accelerated mouse (SAM-P8) brain. J. Neural Transm. 1997, 104, 1267–1275.
- Sureda, F.X.; Gutierrez-Cuesta, J.; Romeu, M.; Mulero, M.; Canudas, A.M.; Camins, A.; Mallol, J.; Pallas, M. Changes in oxidative stress parameters and neurodegeneration markers in the brain of the senescence-accelerated mice SAMP-8. Exp. Gerontol. 2006, 41, 360–367.
- Manich, G.; Mercader, C.; del Valle, J.; Duran-Vilaregut, J.; Camins, A.; Pallàs, M.; Vilaplana, J.; Pelegrí, C. Characterization of amyloid-β granules in the hippocampus of SAMP8 mice. J. Alzheimers Dis. 2011, 25, 535–546.
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38, e100492.
- Alvarez-Garcia, O.; Vega-Naredo, I.; Sierra, V.; Caballero, B.; Tomas-Zapico, C.; Camins, A.; Garcia, J.J.; Pallas, M.; Coto-Montes, A. Elevated oxidative stress in the brain of senescence-accelerated mice at 5 months of age. Biogerontology 2006, 7, 43–52.
- Ota, H.; Akishita, M.; Akiyoshi, T.; Kahyo, T.; Setou, M.; Ogawa, S.; Iijima, K.; Eto, M.; Ouchi, Y. Testosterone deficiency accelerates neuronal and vascular aging of SAMP8 mice: Protective role of eNOS and SIRT1. PLoS ONE 2012, 7, e29598.
- Karuppagounder, V.; Arumugam, S.; Babu, S.S.; Palaniyandi, S.S.; Watanabe, K.; Cooke, J.P.; Thandavarayan, R.A. The senescence accelerated mouse prone 8 (SAMP8): A novel murine model for cardiac aging. Ageing Res. Rev. 2017, 35, 291–296.
- Niimi, K.; Takahashi, E.; Itakura, C. Adiposity-related biochemical phenotype in senescence-accelerated mouse prone 6 (SAMP6). Comp. Med. 2009, 59, 431–436.
- Shimada, A.; Ohta, A.; Akiguchi, I.; Takeda, T. Inbred SAM-P/10 as a mouse model of spontaneous, inherited brain atrophy. J. Neuropathol. Exp. Neurol. 1992, 51, 440–450.
- Shimada, A.; Hosokawa, M.; Ohta, A.; Akiguchi, I.; Takeda, T. Localization of atrophy-prone areas in the aging mouse brain: Comparison between the brain atrophy model SAM-P/10 and the normal control SAM-R/1. Neuroscience 1994, 59, 859–869.
- Shcherbakov, D.; Nigri, M.; Akbergenov, R.; Brilkova, M.; Mantovani, M.; Petit, P.I.; Grimm, A.; Karol, A.A.; Teo, Y.; Sanchón, A.C.; et al. Premature aging in mice with error-prone protein synthesis. Sci. Adv. 2022, 8, eabl9051.
- Brennan, T.A.; Egan, K.P.; Lindborg, C.M.; Chen, Q.; Sweetwyne, M.T.; Hankenson, K.D.; Xie, S.X.; Johnson, F.B.; Pignolo, R.J. Mouse models of telomere dysfunction phenocopy skeletal changes found in human age-related osteoporosis. Dis. Model Mech. 2014, 7, 583–592.
- Botter, S.M.; Zar, M.; van Osch, G.J.; van Steeg, H.; Dolle, M.E.; Hoeijmakers, J.H.; Weinans, H.; van Leeuwen, J.P. Analysis of osteoarthritis in a mouse model of the progeroid human DNA repair syndrome trichothiodystrophy. Age 2011, 33, 247–260.
- Edstrom, E.; Altun, M.; Bergman, E.; Johnson, H.; Kullberg, S.; Ramirez-Leon, V.; Ulfhake, B. Factors contributing to neuromuscular impairment and sarcopenia during aging. Physiol. Behav. 2007, 92, 129–135.
- Baker, D.J.; Jeganathan, K.B.; Cameron, J.D.; Thompson, M.; Juneja, S.; Kopecka, A.; Kumar, R.; Jenkins, R.B.; de Groen, P.C.; Roche, P.; et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 2004, 36, 744–749.
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189.
- Hartman, T.K.; Wengenack, T.M.; Poduslo, J.F.; van Deursen, J.M. Mutant mice with small amounts of BubR1 display accelerated age-related gliosis. Neurobiol. Aging 2007, 28, 921–927.
- Loerch, P.M.; Lu, T.; Dakin, K.A.; Vann, J.M.; Isaacs, A.; Geula, C.; Wang, J.; Pan, Y.; Gabuzda, D.H.; Li, C.; et al. Evolution of the aging brain transcriptome and synaptic regulation. PLoS ONE 2008, 3, e3329.
- Bishop, N.A.; Lu, T.; Yankner, B.A. Neural mechanisms of ageing and cognitive decline. Nature 2010, 464, 529–535.
- Esquerda-Canals, G.; Montoliu-Gaya, L.; Güell-Bosch, J.; Villegas, S. Mouse Models of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1171–1183.
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular Basis of Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 952–963.
- Teich, A.F.; Patel, M.; Arancio, O. A reliable way to detect endogenous murine β-amyloid. PLoS ONE 2013, 8, e55647.
- Myers, A.; McGonigle, P. Overview of Transgenic Mouse Models for Alzheimer’s Disease. Curr. Protoc. Neurosci. 2019, 89, e81.
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Berthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 1995, 373, 523–527.
- Hall, A.M.; Roberson, E.D. Mouse models of Alzheimer’s disease. Brain Res. Bull 2012, 88, 3–12.
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487.
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421.
- Zhang, X.; Luhrs, K.J.; Ryff, K.A.; Malik, W.T.; Driscoll, M.J.; Culver, B. Suppression of nuclear factor kappa B ameliorates astrogliosis but not amyloid burden in APPswe/PS1dE9 mice. Neuroscience 2009, 161, 53–58.
- Sun, X.-Y.; Li, L.-J.; Dong, Q.-X.; Zhu, J.; Huang, Y.-R.; Hou, S.-J.; Yu, X.-L.; Liu, R.-T. Rutin prevents tau pathology and neuroinflammation in a mouse model of Alzheimer’s disease. J. Neuroinflamm. 2021, 18, 131.
- Xie, W.-Q.; He, M.; Yu, D.-J.; Wu, Y.-X.; Wang, X.-H.; Lv, S.; Xiao, W.-F.; Li, Y.-S. Mouse models of sarcopenia: Classification and evaluation. J. Cachexia Sarcopenia Muscle 2021, 12, 538–554.
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693.
- Capitanio, D.; Moriggi, M.; De Palma, S.; Bizzotto, D.; Molon, S.; Torretta, E.; Fania, C.; Bonaldo, P.; Gelfi, C.; Braghetta, P. Collagen VI Null Mice as a Model for Early Onset Muscle Decline in Aging. Front. Mol. Neurosci. 2017, 10, 337.
- Ko, F.; Abadir, P.; Marx, R.; Westbrook, R.; Cooke, C.; Yang, H.; Walston, J. Impaired mitochondrial degradation by autophagy in the skeletal muscle of the aged female interleukin 10 null mouse. Exp. Gerontol. 2016, 73, 23–27.
- Ahn, B.; Smith, N.; Saunders, D.; Ranjit, R.; Kneis, P.; Towner, R.A.; Van Remmen, H. Using MRI to measure in vivo free radical production and perfusion dynamics in a mouse model of elevated oxidative stress and neurogenic atrophy. Redox. Biol. 2019, 26, 101308.
- Sayed, R.K.A.; Fernández-Ortiz, M.; Diaz-Casado, M.E.; Aranda-Martínez, P.; Fernández-Martínez, J.; Guerra-Librero, A.; Escames, G.; López, L.C.; Alsaadawy, R.M.; Acuña-Castroviejo, D. Lack of NLRP3 Inflammasome Activation Reduces Age-Dependent Sarcopenia and Mitochondrial Dysfunction, Favoring the Prophylactic Effect of Melatonin. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2019, 74, 1699–1708.
- Li, J.; Yi, X.; Yao, Z.; Chakkalakal, J.V.; Xing, L.; Boyce, B.F. TNF Receptor-Associated Factor 6 Mediates TNFα-Induced Skeletal Muscle Atrophy in Mice During Aging. J. Bone Min. Res. 2020, 35, 1535–1548.
- Sun, H.; Gong, Y.; Qiu, J.; Chen, Y.; Ding, F.; Zhao, Q. TRAF6 inhibition rescues dexamethasone-induced muscle atrophy. Int. J. Mol. Sci. 2014, 15, 11126–11141.
- Wada, S.; Kato, Y.; Okutsu, M.; Miyaki, S.; Suzuki, K.; Yan, Z.; Schiaffino, S.; Asahara, H.; Ushida, T.; Akimoto, T. Translational suppression of atrophic regulators by microRNA-23a integrates resistance to skeletal muscle atrophy. J. Biol. Chem. 2011, 286, 38456–38465.
- Campos, F.; Abrigo, J.; Aguirre, F.; Garcés, B.; Arrese, M.; Karpen, S.; Cabrera, D.; Andía, M.E.; Simon, F.; Cabello-Verrugio, C. Sarcopenia in a mice model of chronic liver disease: Role of the ubiquitin-proteasome system and oxidative stress. Pflug. Arch. Eur. J. Physiol. 2018, 470, 1503–1519.
- Noll, N.A.; Lal, H.; Merryman, W.D. Mouse Models of Heart Failure with Preserved or Reduced Ejection Fraction. Am. J. Pathol. 2020, 190, 1596–1608.
- Meng, Q.; Lai, Y.C.; Kelly, N.J.; Bueno, M.; Baust, J.J.; Bachman, T.N.; Goncharov, D.; Vanderpool, R.R.; Radder, J.E.; Hu, J.; et al. Development of a Mouse Model of Metabolic Syndrome, Pulmonary Hypertension, and Heart Failure with Preserved Ejection Fraction. Am. J. Respir. Cell Mol. Biol. 2017, 56, 497–505.
- Manrique, C.; DeMarco, V.G.; Aroor, A.R.; Mugerfeld, I.; Garro, M.; Habibi, J.; Hayden, M.R.; Sowers, J.R. Obesity and insulin resistance induce early development of diastolic dysfunction in young female mice fed a Western diet. Endocrinology 2013, 154, 3632–3642.
- Aroor, A.R.; Habibi, J.; Kandikattu, H.K.; Garro-Kacher, M.; Barron, B.; Chen, D.; Hayden, M.R.; Whaley-Connell, A.; Bender, S.B.; Klein, T.; et al. Dipeptidyl peptidase-4 (DPP-4) inhibition with linagliptin reduces western diet-induced myocardial TRAF3IP2 expression, inflammation and fibrosis in female mice. Cardiovasc. Diabetol. 2017, 16, 61.
- Cannon, M.V.; Silljé, H.H.; Sijbesma, J.W.; Khan, M.A.; Steffensen, K.R.; van Gilst, W.H.; de Boer, R.A. LXRα improves myocardial glucose tolerance and reduces cardiac hypertrophy in a mouse model of obesity-induced type 2 diabetes. Diabetologia 2016, 59, 634–643.
- Withaar, C.; Meems, L.M.G.; Markousis-Mavrogenis, G.; Boogerd, C.J.; Silljé, H.H.W.; Schouten, E.M.; Dokter, M.M.; Voors, A.A.; Westenbrink, B.D.; Lam, C.S.P.; et al. The effects of liraglutide and dapagliflozin on cardiac function and structure in a multi-hit mouse model of heart failure with preserved ejection fraction. Cardiovasc. Res. 2021, 117, 2108–2124.
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356.