 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dong-Kug Choi | -- | 2533 | 2022-04-21 02:45:50 | | | |
| 2 | Amina Yu | + 6 word(s) | 2539 | 2022-04-21 06:18:35 | | |
Video Upload Options
Metabotropic glutamate receptors (mGluRs; members of class C G-protein-coupled receptors) have been shown to modulate excitatory neurotransmission, regulate presynaptic extracellular glutamate levels, and modulate postsynaptic ion channels on dendritic spines. mGluRs were found to activate myriad signalling pathways to regulate synapse formation, long-term potentiation, autophagy, apoptosis, necroptosis, and pro-inflammatory cytokines release. A notorious expression pattern of mGluRs has been evident in several neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and schizophrenia. Among the several mGluRs, mGluR5 is one of the most investigated types of considered prospective therapeutic targets and potential diagnostic tools in neurodegenerative diseases and neuropsychiatric disorders.
1. Group I mGluRs in Parkinson’s Disease
1.1. Alterations in Basic Signalling
1.2. Interaction with α-Synuclein
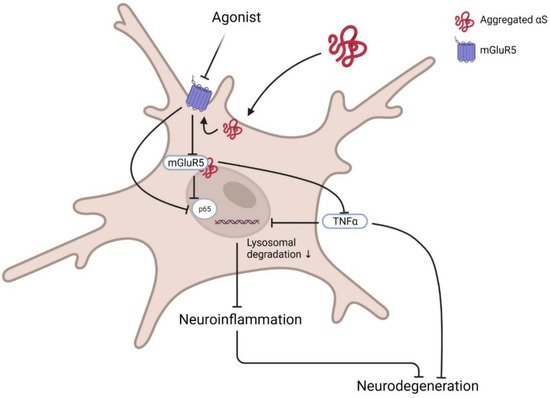
1.3. Modulation of Apoptotic Signalling
2. Neuroimaging of Group I mGluRs for Diagnosis and Therapeutic Development
3. Emerging Therapeutics and Prospective Targets of Group I mGluRs in PD Therapy
3.1. Regulation of Autophagy
3.2. Gut-Brain Axis
References
- Morin, N.; Morissette, M.; Grégoire, L.; Gomez-Mancilla, B.; Gasparini, F.; Di Paolo, T. Chronic treatment with MPEP, an mGlu5 receptor antagonist, normalizes basal ganglia glutamate neurotransmission in l-DOPA-treated parkinsonian monkeys. Neuropharmacology 2013, 73, 216–231.
- Sarantis, K.; Tsiamaki, E.; Kouvaros, S.; Papatheodoropoulos, C.; Angelatou, F. Adenosine A2A receptors permit mGluR5-evoked tyrosine phosphorylation of NR2B (Tyr1472) in rat hippocampus: A possible key mechanism in NMDA receptor modulation. J. Neurochem. 2015, 135, 714–726.
- Crabbé, M.; Van der Perren, A.; Weerasekera, A.; Himmelreich, U.; Baekelandt, V.; Van Laere, K.; Casteels, C. Altered mGluR5 binding potential and glutamine concentration in the 6-OHDA rat model of acute Parkinson’s disease and levodopa-induced dyskinesia. Neurobiol. Aging 2018, 61, 82–92.
- Marques, O.; Outeiro, T.F. Alpha-synuclein: From secretion to dysfunction and death. Cell Death Dis. 2012, 3, e350.
- Ferreira, D.G.; Ferreira, M.T.; Miranda, H.V.; Batalha, V.L.; Coelho, J.; Szegö, É.M.; Marques-Morgado, I.; Vaz, S.H.; Rhee, J.S.; Schmitz, M.; et al. α-synuclein interacts with PrPC to induce cognitive impairment through mGluR5 and NMDAR2B. Nat. Neurosci. 2017, 20, 1569–1579.
- Zhang, Y.-N.; Fan, J.-K.; Gu, L.; Yang, H.-M.; Zhan, S.-Q.; Zhang, H. Metabotropic glutamate receptor 5 inhibits α-synuclein-induced microglia inflammation to protect from neurotoxicity in Parkinson’s disease. J. Neuroinflammation 2021, 18, 23.
- Beraldo, F.H.; Ostapchenko, V.; Caetano, F.A.; Guimaraes, A.; Ferretti, G.D.S.; Daude, N.; Bertram, L.; Nogueira, K.O.P.C.; Silva, J.; Westaway, D.; et al. Regulation of Amyloid β Oligomer Binding to Neurons and Neurotoxicity by the Prion Protein-mGluR5 Complex. J. Biol. Chem. 2016, 291, 21945–21955.
- Resenberger, U.K.; Harmeier, A.; Woerner, A.C.; Goodman, J.L.; Müller, V.; Krishnan, R.; Vabulas, R.M.; Kretzschmar, H.A.; Lindquist, S.; Hartl, F.U.; et al. The cellular prion protein mediates neurotoxic signalling of β-sheet-rich conformers independent of prion replication. EMBO J. 2011, 30, 2057–2070.
- Jansson, L.C.; Åkerman, K.E. The role of glutamate and its receptors in the proliferation, migration, differentiation and survival of neural progenitor cells. J. Neural Transm. 2014, 121, 819–836.
- Copani, A.; Casabona, G.; Bruno, V.; Caruso, A.; Condorelli, D.-F.; Messina, A.; Gerevini, V.D.G.; Pin, J.-P.; Kuhn, R.; Knöpfel, T.; et al. The metabotropic glutamate receptor mGlu5 controls the onset of developmental apoptosis in cultured cerebellar neurons. Eur. J. Neurosci. 1998, 10, 2173–2184.
- Ulus, I.H.; Wurtman, R.J. Metabotropic glutamate receptor agonists increase release of soluble amyloid precursor protein derivatives from rat brain cortical and hippocampal slices. J. Pharmacol. Exp. Ther. 1997, 281, 149–154.
- Luo, W.Y.; Xing, S.Q.; Zhu, P.; Zhang, C.G.; Yang, H.M.; Van Halm-Lutterodt, N.; Gu, L.; Zhang, H. PDZ Scaffold Protein CAL Couples with Metabotropic Glutamate Receptor 5 to Protect Against Cell Apoptosis and Is a Potential Target in the Treatment of Parkinson’s Disease. Neurotherapeutics 2019, 16, 761–783.
- Peng, J.; Andersen, J. The Role of c-Jun N-Terminal Kinase (JNK) in Parkinson’s Disease. IUBMB Life 2003, 55, 267–271.
- Paquet, M.; Ribeiro, F.M.; Guadagno, J.; Esseltine, J.L.; Ferguson, S.S.; Cregan, S.P. Role of metabotropic glutamate receptor 5 signaling and homer in oxygen glucose deprivation-mediated astrocyte apoptosis. Mol. Brain 2013, 6, 9.
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73.
- Proskuryakov, S.; Gabai, S.Y.P.A.V.L. Mechanisms of Tumor Cell Necrosis. Curr. Pharm. Des. 2010, 16, 56–68.
- Jantas, D.; Greda, A.; Golda, S.; Korostynski, M.; Grygier, B.; Roman, A.; Pilc, A.; Lason, W. Neuroprotective effects of metabotropic glutamate receptor group II and III activators against MPP(+)-induced cell death in human neuroblastoma SH-SY5Y cells: The impact of cell differentiation state. Neuropharmacology 2014, 83, 36–53.
- Caraci, F.; Battaglia, G.; Sortino, M.A.; Spampinato, S.F.; Molinaro, G.; Copani, A.; Nicoletti, F.; Bruno, V.M.G. Metabotropic glutamate receptors in neurodegeneration/neuroprotection: Still a hot topic? Neurochem. Int. 2012, 61, 559–565.
- Nicoletti, F.; Bockaert, J.; Collingridge, G.; Conn, P.; Ferraguti, F.; Schoepp, D.; Wroblewski, J.; Pin, J.-P. Metabotropic glutamate receptors: From the workbench to the bedside. Neuropharmacology 2011, 60, 1017–1041.
- Holmes, S.E.; Gallezot, J.-D.; Davis, M.T.; DellaGioia, N.; Matuskey, D.; Nabulsi, N.; Krystal, J.H.; Javitch, J.A.; DeLorenzo, C.; Carson, R.E.; et al. Measuring the effects of ketamine on mGluR5 using FPEB and PET. J. Cereb. Blood Flow Metab. 2019, 40, 2254–2264.
- Kang, Y.; Henchcliffe, C.; Verma, A.; Vallabhajosula, S.; He, B.; Kothari, P.J.; Pryor, K.; Mozley, P.D. 18F-FPEB PET/CT Shows mGluR5 Upregulation in Parkinson’s Disease. J. Neuroimaging 2018, 29, 97–103.
- Varnäs, K.; Juréus, A.; Finnema, S.J.; Johnström, P.; Raboisson, P.; Amini, N.; Takano, A.; Stepanov, V.; Halldin, C.; Farde, L. The metabotropic glutamate receptor 5 radioligand AZD9272 identifies unique binding sites in primate brain. Neuropharmacology 2018, 135, 455–463.
- Amalric, M. Targeting metabotropic glutamate receptors (mGluRs) in Parkinson’s disease. Curr. Opin. Pharmacol. 2015, 20, 29–34.
- Rylander, D.; Recchia, A.; Mela, F.; Dekundy, A.; Danysz, W.; Cenci, M.A. Pharmacological Modulation of Glutamate Transmission in a Rat Model of l-DOPA-Induced Dyskinesia: Effects on Motor Behavior and Striatal Nuclear Signaling. J. Pharmacol. Exp. Ther. 2009, 330, 227–235.
- Litim, N.; Morissette, M.; Di Paolo, T. Metabotropic glutamate receptors as therapeutic targets in Parkinson’s disease: An update from the last 5 years of research. Neuropharmacology 2017, 115, 166–179.
- Ribeiro, F.; Vieira, L.B.; Pires, R.G.; Olmo, R.P.; Ferguson, S.S. Metabotropic glutamate receptors and neurodegenerative diseases. Pharmacol. Res. 2017, 115, 179–191.
- Varnäs, K.; Cselényi, Z.; Arakawa, R.; Nag, S.; Stepanov, V.; Moein, M.M.; Johnström, P.; Kingston, L.; Elmore, C.; Halldin, C.; et al. The pro-psychotic metabotropic glutamate receptor compounds fenobam and AZD9272 share binding sites with monoamine oxidase-B inhibitors in humans. Neuropharmacology 2020, 162, 107809.
- Ambrosi, G.; Armentero, M.-T.; Levandis, G.; Bramanti, P.; Nappi, G.; Blandini, F. Effects of early and delayed treatment with an mGluR5 antagonist on motor impairment, nigrostriatal damage and neuroinflammation in a rodent model of Parkinson’s disease. Brain Res. Bull. 2010, 82, 29–38.
- Morin, N.; Grégoire, L.; Gomez-Mancilla, B.; Gasparini, F.; Di Paolo, T. Effect of the metabotropic glutamate receptor type 5 antagonists MPEP and MTEP in parkinsonian monkeys. Neuropharmacology 2010, 58, 981–986.
- Morin, N.; Grégoire, L.; Morissette, M.; Desrayaud, S.; Gomez-Mancilla, B.; Gasparini, F.; Di Paolo, T. MPEP, an mGlu5 receptor antagonist, reduces the development of l-DOPA-induced motor complications in de novo parkinsonian monkeys: Biochemical correlates. Neuropharmacology 2013, 66, 355–364.
- Maranis, S.; Stamatis, D.; Tsironis, C.; Konitsiotis, S. Investigation of the antidyskinetic site of action of metabotropic and ionotropic glutamate receptor antagonists. Intracerebral infusions in 6-hydroxydopamine-lesioned rats with levodopa-induced dyskinesia. Eur. J. Pharmacol. 2012, 683, 71–77.
- Grégoire, L.; Morin, N.; Ouattara, B.; Gasparini, F.; Bilbe, G.; Johns, D.; Vranesic, I.; Sahasranaman, S.; Gomez-Mancilla, B.; Di Paolo, T. The acute antiparkinsonian and antidyskinetic effect of AFQ056, a novel metabotropic glutamate receptor type 5 antagonist, in l-Dopa-treated parkinsonian monkeys. Park. Relat. Disord. 2011, 17, 270–276.
- Bezard, E.; Pioli, E.Y.; Li, Q.; Girard, F.; Mutel, V.; Keywood, C.; Tison, F.; Rascol, O.; Poli, S.M. The mGluR5 negative allosteric modulator dipraglurant reduces dyskinesia in the MPTP macaque model. Mov. Disord. 2014, 29, 1074–1079.
- Ko, W.K.D.; Pioli, E.; Li, Q.; McGuire, S.; Dufour, A.; Sherer, T.B.; Bezard, E.; Facheris, M.F. Combined fenobam and amantadine treatment promotes robust antidyskinetic effects in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-lesioned primate model of Parkinson’s disease. Mov. Disord. 2014, 29, 772–779.
- Chen, L.; Liu, J.; Ali, U.; Gui, Z.H.; Hou, C.; Fan, L.L.; Wang, Y.; Wang, T. Chronic, systemic treatment with a metabotropic glutamate receptor 5 antagonist produces anxiolytic-like effects and reverses abnormal firing activity of projection neurons in the basolateral nucleus of the amygdala in rats with bilateral 6-OHDA lesions. Brain Res. Bull. 2011, 84, 215–223.
- Hsieh, M.-H.; Ho, S.-C.; Yeh, K.-Y.; Pawlak, C.R.; Chang, H.-M.; Ho, Y.-J.; Lai, T.-J.; Wu, F.-Y. Blockade of metabotropic glutamate receptors inhibits cognition and neurodegeneration in an MPTP-induced Parkinson’s disease rat model. Pharmacol. Biochem. Behav. 2012, 102, 64–71.
- Fieblinger, T.; Sebastianutto, I.; Alcacer, C.; Bimpisidis, Z.; Maslava, N.; Sandberg, S.; Engblom, D.; Cenci, M.A. Mechanisms of Dopamine D1 Receptor-Mediated ERK1/2 Activation in the Parkinsonian Striatum and Their Modulation by Metabotropic Glutamate Receptor Type. J. Neurosci. 2014, 34, 4728–4740.
- Masilamoni, G.J.; Smith, Y. Metabotropic glutamate receptors: Targets for neuroprotective therapies in Parkinson disease. Curr. Opin. Pharmacol. 2018, 38, 72–80.
- Ibrahim, K.S.; McLaren, C.J.; Abd-Elrahman, K.S.; Ferguson, S.S. Optineurin deletion disrupts metabotropic glutamate receptor 5-mediated regulation of ERK1/2, GSK3β/ZBTB16, mTOR/ULK1 signaling in autophagy. Biochem. Pharmacol. 2021, 185, 114427.
- Abd-Elrahman, K.S.; Hamilton, A.; Hutchinson, S.R.; Liu, F.; Russell, R.C.; Ferguson, S.S.G. mGluR5 antagonism increases autophagy and prevents disease progression in the zQ175 mouse model of Huntington’s disease. Sci. Signal. 2017, 10, aan6387.
- Abd-Elrahman, K.S.; Ferguson, S.S.G. Modulation of mTOR and CREB pathways following mGluR5 blockade contribute to improved Huntington’s pathology in zQ175 mice. Mol. Brain 2019, 12, 35.
- Abd-Elrahman, K.S.; Hamilton, A.; Albaker, A.; Ferguson, S.S.G. mGluR5 Contribution to Neuropathology in Alzheimer Mice Is Disease Stage-Dependent. ACS Pharmacol. Transl. Sci. 2020, 3, 334–344.
- Niu, Y.; Zeng, X.; Qin, G.; Zhang, D.; Zhou, J.; Chen, L. Downregulation of metabotropic glutamate receptor 5 alleviates central sensitization by activating autophagy via inhibiting mTOR pathway in a rat model of chronic migraine. Neurosci. Lett. 2021, 743, 135552.
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211.
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.
- Young, R.L.; Page, A.J.; O’Donnell, T.A.; Cooper, N.J.; Blackshaw, L.A.; Blackshaw, A. Peripheral versus central modulation of gastric vagal pathways by metabotropic glutamate receptor Am. J. Physiol. Liver Physiol. 2007, 292, G501–G511.

