As mGluR1 and mGluR5 are widely expressed in the basal ganglia structures, especially at postsynaptic sites [
90], and a high expression of mGluR1 receptors can be found in the globus pallidum (GP), substantia nigra pars reticulata (SNr), and striatum, therefore they might be involved in PD pathogenesis. A study showed that antagonism of mGluR1 using negative allosteric modulators (NAMs) did not reduce LID in PD; only blocking of mGluR5 showed a promising reduction of dyskinesia [
91]. Some studies also showed that using the mGluR5 NAMs. such as 2-methyl-6-(phenylethynyl)-pyridine (MPEP), mavoglurant, dipraglurant, fenobam, and 3-((2-Methyl-4-thiazolyl)ethynyl)pyridine (MTEP), were shown to ameliorate motor deficits in PD animals [
8,
91,
92]. Fenobam and AZD9272 have been reported to induce psychosis-like adverse events. Varnas et al. (2020) reported from a PET study of the human brain that both antagonists bind to monoamine oxidase-B (MAO-B), which reveals a new understanding of psychosis-like adverse effects and could generate new models for the pathophysiology of psychosis [
93]. MPEP treatment significantly ameliorated akinesia in 6-hydroxydopamine (6-OHDA)-induced rodents and decreased LID in MPTP treated monkeys [
94,
95]. Chronic treatment with MPEP in MPTP-treated PD monkeys for 1 month was found to inhibit LID [
96]. Administration of MTEP also showed a significant decrease of dyskinesia in MPTP-treated monkeys [
95] and 6-OHDA-lesioned rats [
97]. Several studies with different other NAMs such as mavoglurant [
98], dipraglurant [
99], and fenobam [
100] also reported similar anti-parkinsonism and a decrease in LID of L-DOPA in different PD models. MPEP chronic treatment was shown to attenuate DA neuronal loss and prevented microglial activation in SNpc of 6-OHDA treated or MPTP-treated rats [
101,
102]. Moreover, MTEP local infusion in the striatum was reported to attenuate 6-OHDA-induced activation of ERK1/2 signalling that is associated with dyskinesia [
103]. Different antagonists of mGluR5, including AFQ056 (mavoglurant) and ADX-48621 (dipraglurant), are currently being tested in humans as anti-dyskinetic drugs. These drugs are well tolerated and have still not been reported to worsen PD motor symptoms [
104], which is encouraging and supportive to study further and develop mGluR5-related compounds as potential neuroprotective drugs in PD.
3.1. Regulation of Autophagy
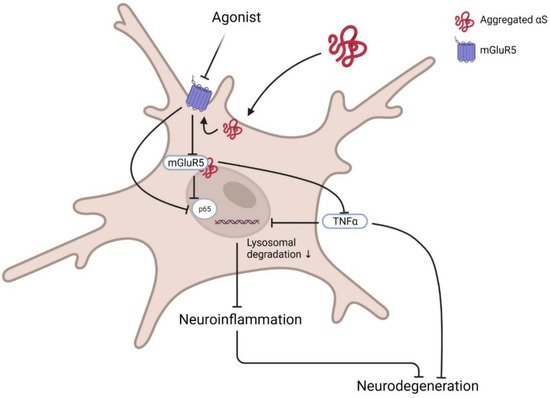
Group I mGluRs are potential regulators of several autophagic signal transducers that contribute to the pathophysiology of neurodegenerative diseases such as AD and HD. However, the role of members of this mGluR group has not been evaluated yet in the PD model; hence, in this section, we prospected a few potential targets that might interest mGluR-mediated autophagy-based therapeutic development. Optineurin is a multifunctional cellular network processor protein that regulates membrane trafficking, inflammatory response, and autophagy, and mGluR5-mediated autophagic signalling is regulated by optineurin [
105]. mGluR5 couples to canonical Gαq/11 and activates autophagic machinery via mTOR/ULK1 and GSK3β/ZBTB16 pathways. In this process, mGluR5 promotes intracellular Ca
2+-influx and signals ERK1/2; interestingly, optineurin regulates mGluR5-mediated Ca
2+ and mTOR/ULK1 and GSK3β/ZBTB16 pathway activation. Although crosstalk between optineurin and mGluR5 and their contribution to neurodegenerative diseases pathology is now known [
105], downstream signalling remains largely unknown.
In addition, long-term use of mGluR5 NAM (CTEP) attenuated caspase-3 activation, neuronal loss, and apoptosis in both heterozygous and homozygous knock-in HD mice models [
106], which occurred via activation of GSK3β/ZBTB16-mediated autophagy. Inhibition of mGluR5 attenuates autophagosome biogenesis-related kinase ULK1 and increases autophagy factor ATG13 and Beclin1. In addition, inhibition of mGluR5 by CTEP reduces aberrant phosphorylation of the PI3K/Akt/mTOR signalling cascade [
107] that promotes ULK1 activity and autophagy. Antagonism of mGluR5 via selective NAM (CTEP) promotes aggregated protein clearance by autophagy activation and facilitates CREB-mediated BDNF expression in the brain, fostering neuronal survival and reducing apoptosis. In addition, chronic use of CTEP for 24 weeks was shown to reduce the Aβ burden in APPswe/PS1ΔE9 mouse hippocampus and cortex; however, CTEP at 36 weeks became ineffective [
108]. Reflecting that mutation at APP in the advanced disease stage could alter mGluR5 expression and mGluR5-mediated ZBTB16 and mTOR signalling in the brain. Inhibition of mGluR5 and subsequent mTOR phosphorylation could also alleviate inflammatory responses by decreasing IL-1β expression that might have been correlated with the activation of autophagy [
109].