 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mario Cappiello | -- | 3013 | 2022-04-11 12:16:25 | | | |
| 2 | Rita Xu | Meta information modification | 3013 | 2022-04-11 12:25:27 | | |
Video Upload Options
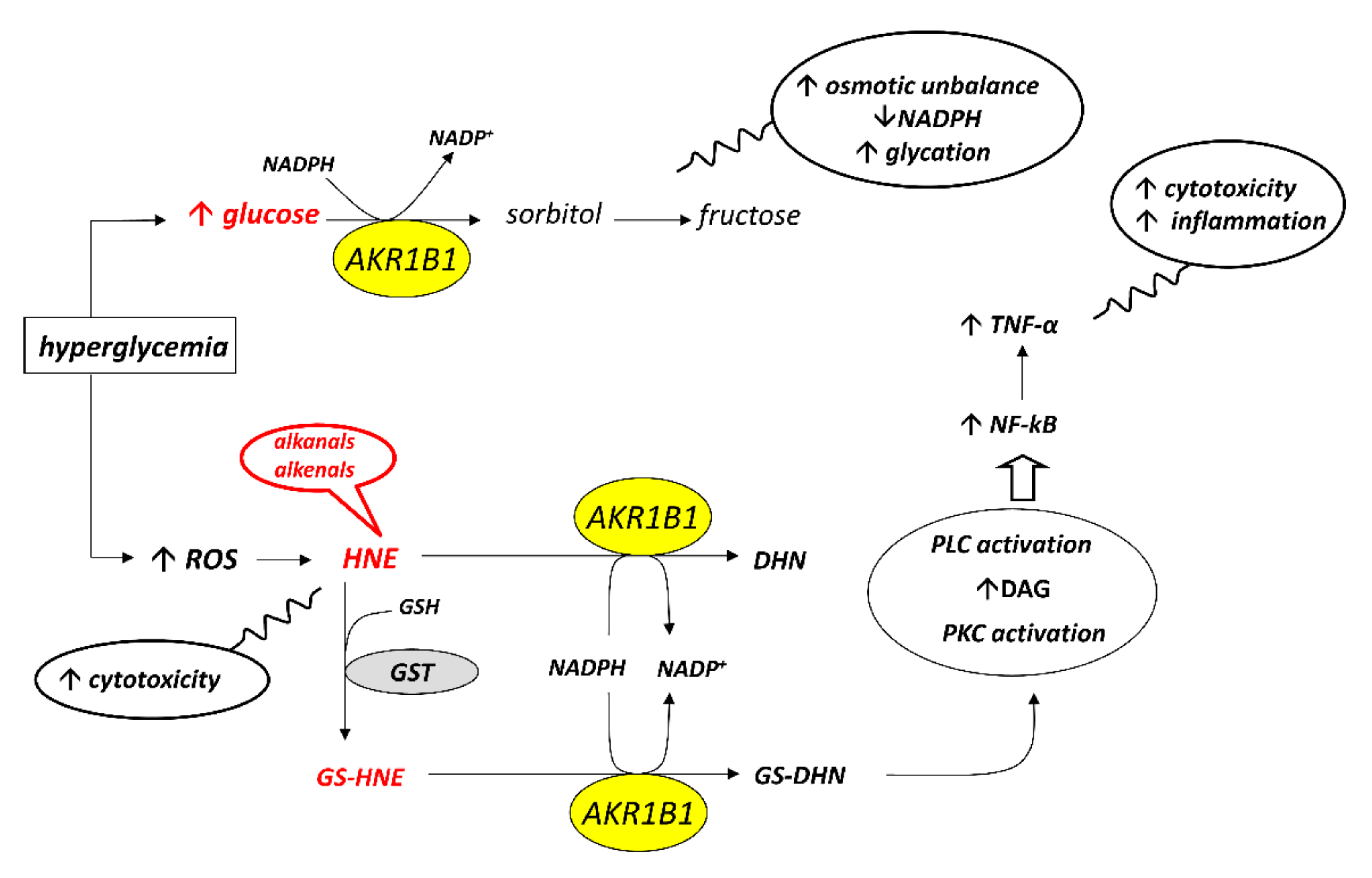
Aldose reductase, classified within the aldo-keto reductase family as AKR1B1, is an NADPH dependent enzyme that catalyzes the reduction of hydrophilic as well as hydrophobic aldehydes. AKR1B1 is the first enzyme of the so-called polyol pathway that allows the conversion of glucose into sorbitol, which in turn is oxidized to fructose by sorbitol dehydrogenase. The activation of the polyol pathway in hyperglycemic conditions is generally accepted as the event that is responsible for a series of long-term complications of diabetes such as retinopathy, cataract, nephropathy and neuropathy. The role of AKR1B1 in the onset of diabetic complications has made this enzyme the target for the development of molecules capable of inhibiting its activity.
1. Introduction
2. Aldose Reductase and the Polyol Pathway
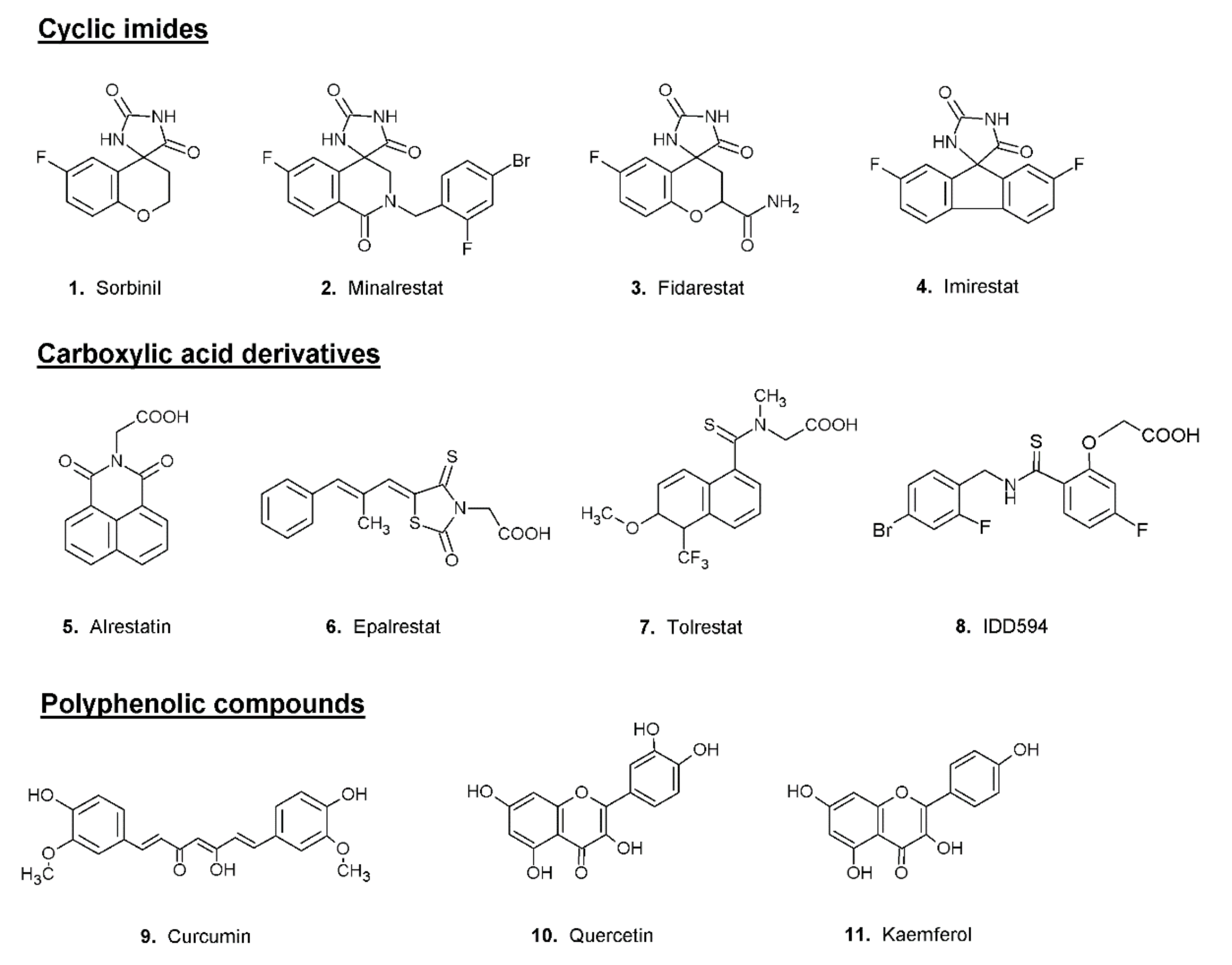
3. Aldose Reductase Inhibitors
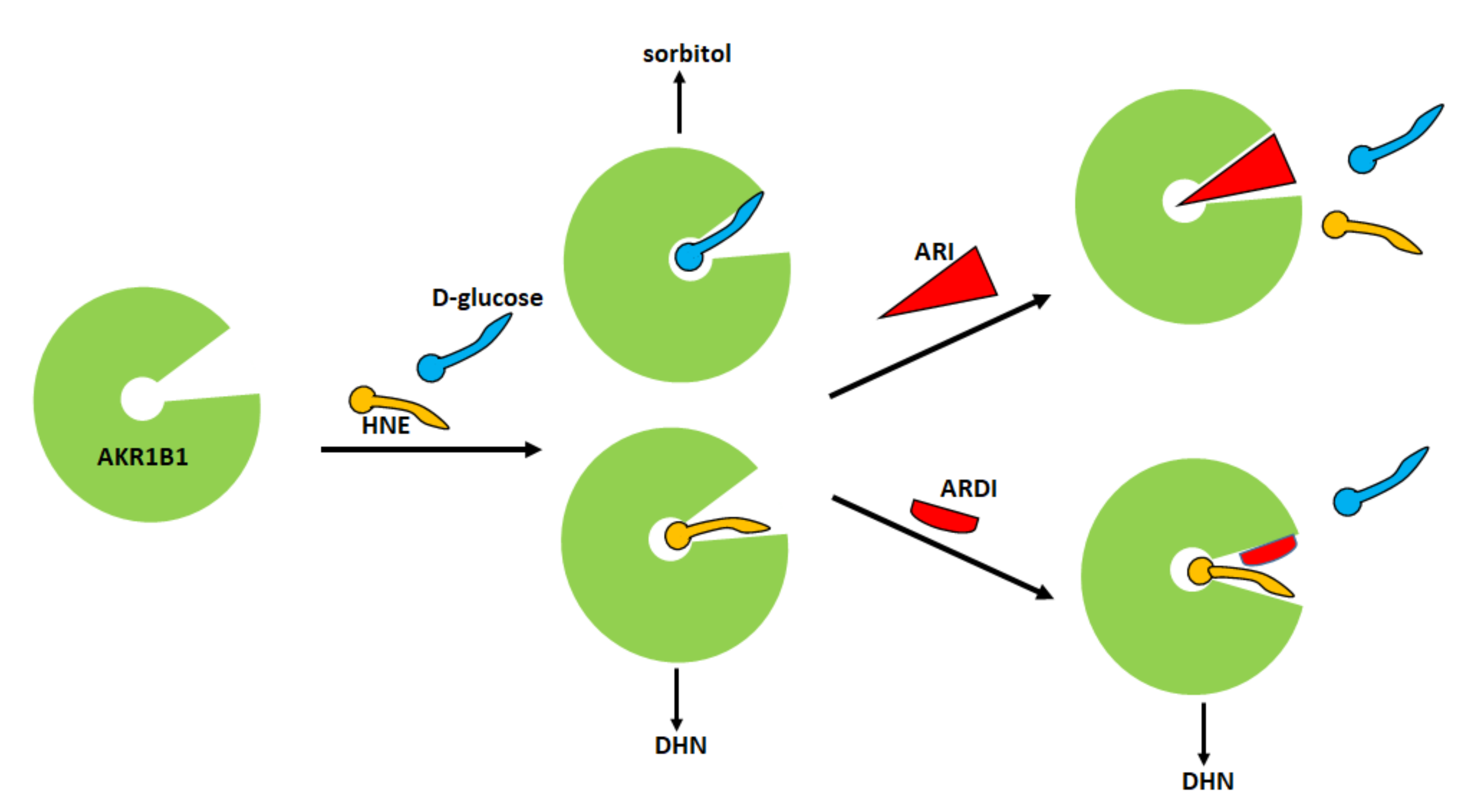
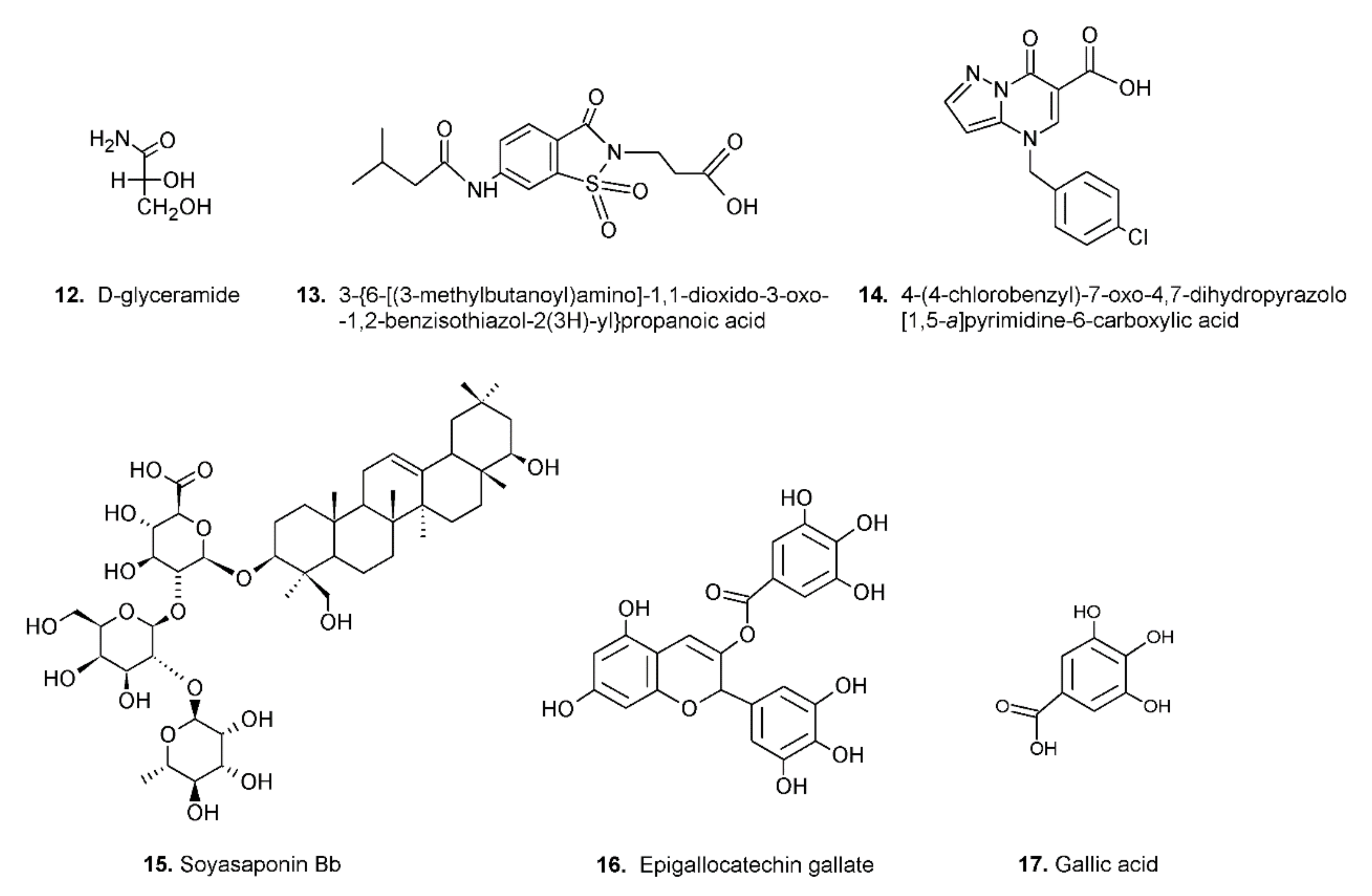
4. Aldose Reductase Differential Inhibitors
| Hydrophobic Substrates and Derivatives | Ks mM−1min−1 |
Aldoses | Ks mM−1min−1 |
|---|---|---|---|
| propanal | 5.5 | d-glyceraldehyde | 1860 |
| butanal | 439 | l-glyceraldehyde | 6840 |
| hexanal | 3657 | d-threose | 114 |
| nonanal | 1157 | l-threose | 276 |
| trans-2-pentenal | 61.7 | d-arabinose | 0.24 |
| trans-2-nonenal | 2058 | l-arabinose | 10.8 |
| 4-hydroxy trans-2-pentenal | 199 | d-xylose | 8.4 |
| 4-hydroxy trans-2-nonenal | 921 | l-xylose | 0.48 |
| trans-4-decenal | 1108 | d-idose | 4.2 |
| cis-4-decenal | 862 | l-idose | 35.75 |
| 4-hydroxy trans-2,3-nonenal | 2320 | d-glucose | 0.61 |
| 3-glutathionyl-4-hydroxy-nonanal | 1376 | l-glucose | n.d. b |
References
- Penning, T.M. The aldo-keto reductases (AKRs): Overview. Chem.-Biol. Interact. 2015, 234, 236–246.
- Tanimoto, T.; Maekawa, K.; Okada, S.; Yabe-Nishimura, C. Clinical analysis of aldose reductase for differential diagnosis of the pathogenesis of diabetic complication. Anal. Chim. Acta 1998, 365, 285–292.
- Del Corso, A.; Cappiello, M.; Mura, U. From a dull enzyme to something else: Facts and perspectives regarding aldose reductase. Curr. Med. Chem. 2008, 15, 1452–1461.
- Barski, O.A.; Tipparaju, S.M.; Bhatnagar, A. The Aldo-Keto Reductase Superfamily and Its Role in Drug Metabolism and Detoxification. Drug Metab. Rev. 2008, 40, 553–624.
- Bohren, K.M.; Brownlee, J.M.; Milne, A.C.; Gabbay, K.H.; Harrison, D.H.T. The structure of Apo R268A human aldose reductase: Hinges and latches that control the kinetic mechanism. Biochim. Biophys. Acta 2005, 1748, 201–212.
- Harrison, D.H.; Bohren, K.M.; Ringe, D.; Petsko, G.A.; Gabbay, K.H. An anion binding site in human aldose reductase: Mechanistic implications for the binding of citrate, cacodylate, and glucose 6-phosphate. Biochemistry 1994, 33, 2011–2020.
- Urzhumtsev, A.; Tete-Favier, F.; Mitschler, A.; Barbanton, J.; Barth, P.; Urzhumtseva, L.; Biellmann, J.F.; Podjarny, A.; Moras, D. A ‘specificity’ pocket inferred from the crystal structures of the complexes of aldose reductase with the pharmaceutically important inhibitors tolrestat and sorbinil. Structure 1997, 5, 601–612.
- Klebe, G.; Kramer, O.; Sotriffer, C. Strategies for the design of inhibitors of aldose reductase, an enzyme showing pronounced induced-fit adaptations. Cell Mol. Life Sci. 2004, 61, 783–793.
- Sotriffer, C.A.; Kramer, O.; Klebe, G. Probing flexibility and “induced-fit” phenomena in aldose reductase by comparative crystal structure analysis and molecular dynamics simulations. Proteins 2004, 56, 52–66.
- Hymavati; Kumar, V.; Sobhia, M.E. Implication of Crystal Water Molecules in Inhibitor Binding at ALR2 Active Site. Comput. Math. Methods Med. 2012, 2012, 541594.
- Sandner, A.; Ngo, K.; Sager, C.P.; Scheer, F.; Daude, M.; Diederich, W.E.; Heine, A.; Klebe, G. Which Properties Allow Ligands to Open and Bind to the Transient Binding Pocket of Human Aldose Reductase? Biomolecules 2021, 11, 1837.
- Toth, E.; Racz, A.; Toth, J.; Kaminski, P.M.; Wolin, M.S.; Bagi, Z.; Koller, A. Contribution of polyol pathway to arteriolar dysfunction in hyperglycemia. Role of oxidative stress, reduced NO, and enhanced PGH(2)/TXA(2) mediation. Am. J. Physiol.-Heart Circ. Physiol. 2007, 293, H3096–H3104.
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42.
- Stevens, M.J.; Dananberg, J.; Feldman, E.L.; Lattimer, S.A.; Kamijo, M.; Thomas, T.P.; Shindo, H.; Sima, A.A.F.; Greene, D.A. The Linked Roles of Nitric-Oxide, Aldose Reductase and, (Na+,K+)-Atpase in the Slowing of Nerve-Conduction in the Streptozotocin-Diabetic Rat. J. Clin. Investig. 1994, 94, 853–859.
- Goldsmith, Z.G.; Dhanasekaran, D.N. G protein regulation of MAPK networks. Oncogene 2007, 26, 3122–3142.
- Geraldes, P.; King, G.L. Activation of Protein Kinase C Isoforms and Its Impact on Diabetic Complications. Circ. Res. 2010, 106, 1319–1331.
- Busch, M.; Franke, S.; Ruster, C.; Wolf, G. Advanced glycation end-products and the kidney. Eur. J. Clin. Investig. 2010, 40, 742–755.
- Gugliucci, A. Formation of Fructose-Mediated Advanced Glycation End Products and Their Roles in Metabolic and Inflammatory Diseases. Adv. Nutr. 2017, 8, 54–62.
- Hou, B.Y.; Qiang, G.F.; Zhao, Y.R.; Yang, X.Y.; Chen, X.; Yan, Y.; Wang, X.B.; Liu, C.; Zhang, L.; Du, G.H. Salvianolic Acid A Protects Against Diabetic Nephropathy through Ameliorating Glomerular Endothelial Dysfunction via Inhibiting AGE-RAGE Signaling. Cell Physiol. Biochem. 2017, 44, 2378–2394.
- Ramana, K.V.; Srivastava, S.K. Aldose reductase: A novel therapeutic target for inflammatory pathologies. Int. J. Biochem. Cell Biol. 2010, 42, 17–20.
- Wilson, D.K.; Tarle, I.; Petrash, J.M.; Quiocho, F.A. Refined 1.8 A structure of human aldose reductase complexed with the potent inhibitor zopolrestat. Proc. Natl. Acad. Sci. USA 1993, 90, 9847–9851.
- Sarges, R.; Oates, P.J. Aldose reductase inhibitors: Recent developments. Prog. Drug Res. 1993, 40, 99–161.
- Harrison, D.H.T.; Bohren, K.M.; Petsko, G.A.; Ringe, D.; Gabbay, K.H. The Alrestatin double-decker: Binding of two inhibitor molecules to human aldose reductase reveals a new specificity determinant. Biochemistry 1997, 36, 16134–16140.
- Datiles, M.B., 3rd; Fukui, H. Cataract prevention in diabetic Octodon degus with Pfizer’s sorbinil. Curr. Eye Res. 1989, 8, 233–237.
- Fagius, J.; Brattberg, A.; Jameson, S.; Berne, C. Limited benefit of treatment of diabetic polyneuropathy with an aldose reductase inhibitor: A 24-week controlled trial. Diabetologia 1985, 28, 323–329.
- Spielberg, S.P.; Shear, N.H.; Cannon, M.; Hutson, N.J.; Gunderson, K. In-vitro assessment of a hypersensitivity syndrome associated with sorbinil. Ann. Intern. Med. 1991, 114, 720–724.
- Grewal, A.S.; Bhardwaj, S.; Pandita, D.; Lather, V.; Sekhon, B.S. Updates on Aldose Reductase Inhibitors for Management of Diabetic Complications and Non-diabetic Diseases. Mini Rev. Med. Chem. 2016, 16, 120–162.
- He, J.; Gao, H.X.; Yang, N.; Zhu, X.D.; Sun, R.B.; Xie, Y.; Zeng, C.H.; Zhang, J.W.; Wang, J.K.; Ding, F.; et al. The aldose reductase inhibitor epalrestat exerts nephritic protection on diabetic nephropathy in db/db mice through metabolic modulation. Acta Pharmacol. Sin. 2019, 40, 86–97.
- Sestanj, K.; Bellini, F.; Fung, S.; Abraham, N.; Treasurywala, A.; Humber, L.; Simardduquesne, N.; Dvornik, D. N-Thioxomethyl]-N-Methylglycine (Tolrestat), a Potent, Orally Active Aldose Reductase Inhibitor. J. Med. Chem. 1984, 27, 255–256.
- Boulton, A.J.; Levin, S.; Comstock, J. A multicentre trial of the aldose-reductase inhibitor, tolrestat, in patients with symptomatic diabetic neuropathy. Diabetologia 1990, 33, 431–437.
- Santiago, J.V.; Snksen, P.H.; Boulton, A.J.; Macleod, A.; Beg, M.; Bochenek, W.; Graepel, G.J.; Gonen, B. Withdrawal of the aldose reductase inhibitor tolrestat in patients with diabetic neuropathy: Effect on nerve function. The Tolrestat Study Group. J. Diabetes Complicat. 1993, 7, 170–178.
- Grewal, A.S.; Thapa, K.; Kanojia, N.; Sharma, N.; Singh, S. Natural Compounds as Source of Aldose Reductase (AR) Inhibitors for the Treatment of Diabetic Complications: A Mini Review. Curr. Drug Metab. 2020, 21, 1091–1116.
- Howard, E.I.; Sanishvili, R.; Cachau, R.E.; Mitschler, A.; Chevrier, B.; Barth, P.; Lamour, V.; Van Zandt, M.; Sibley, E.; Bon, C.; et al. Ultrahigh resolution drug design I: Details of interactions in human aldose reductase-inhibitor complex at 0.66 angstrom. Proteins 2004, 55, 792–804.
- Steuber, H.; Czodrowski, P.; Sotriffer, C.A.; Klebe, G. Tracing changes in protonation: A prerequisite to factorize thermodynamic data of inhibitor binding to aldose reductase. J. Mol. Biol. 2007, 373, 1305–1320.
- Veeresham, C.; Rao, A.R.; Asres, K. Aldose Reductase Inhibitors of Plant Origin. Phytother. Res. 2014, 28, 317–333.
- Chatzopoulou, M.; Alexiou, P.; Kotsampasakou, E.; Demopoulos, V.J. Novel aldose reductase inhibitors: A patent survey (2006–present). Expert Opin. Ther. Pat. 2012, 22, 1303–1323.
- Imran, A.; Shehzad, M.T.; al Adhami, T.; Rahman, K.M.; Hussain, D.; Alharthy, R.D.; Shafiq, Z.; Iqbal, J. Development of coumarin-thiosemicarbazone hybrids as aldose reductase inhibitors: Biological assays, molecular docking, simulation studies and ADME evaluation. Bioorg. Chem. 2021, 115, 105164.
- Kovacikova, L.; Prnova, M.S.; Majekova, M.; Bohac, A.; Karasu, C.; Stefek, M. Development of Novel Indole-Based Bifunctional Aldose Reductase Inhibitors/Antioxidants as Promising Drugs for the Treatment of Diabetic Complications. Molecules 2021, 26, 2867.
- Zuo, G.; Je, K.H.; Quispe, Y.G.N.; Shin, K.O.; Kim, H.Y.; Kim, K.H.; Arce, P.H.G.; Lim, S.S. Separation and Identification of Antioxidants and Aldose Reductase Inhibitors in Lepechinia meyenii (Walp.) Epling. Plants 2021, 10, 2773.
- Dewangan, D.; Vaishnav, Y.; Mishra, A.; Jha, A.K.; Verma, S.; Badwaik, H. Synthesis, molecular docking, and biological evaluation of Schiff base hybrids of 1,2,4-triazole-pyridine as dihydrofolate reductase inhibitors. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100024.
- Del-Corso, A.; Balestri, F.; Di Bugno, E.; Moschini, R.; Cappiello, M.; Sartini, S.; La-Motta, C.; Da-Settimo, F.; Mura, U. A New Approach to Control the Enigmatic Activity of Aldose Reductase. PLoS ONE 2013, 8, e74076.
- Cappiello, M.; Voltarelli, M.; Giannessi, M.; Cecconi, I.; Camici, G.; Manao, G.; Delcorso, A.; Mura, U. Glutathione-Dependent Modification of Bovine Lens Aldose Reductase. Exp. Eye Res. 1994, 58, 491–501.
- Grimshaw, C.E. Aldose reductase: Model for a new paradigm of enzymic perfection in detoxification catalysts. Biochemistry 1992, 31, 10139–10145.
- Grimshaw, C.E.; Shahbaz, M.; Putney, C.G. Mechanistic Basis for Nonlinear Kinetics of Aldehyde Reduction Catalyzed by Aldose Reductase. Biochemistry 1990, 29, 9947–9955.
- Balestri, F.; Cappiello, M.; Moschini, R.; Rotondo, R.; Abate, M.; Del-Corso, A.; Mura, U. Modulation of aldose reductase activity by aldose hemiacetals. Biochim. Biophys. Acta 2015, 1850, 2329–2339.
- Balestri, F.; Sorce, C.; Moschini, R.; Cappiello, M.; Misuri, L.; Del Corso, A.; Mura, U. Edible vegetables as a source of aldose reductase differential inhibitors. Chem.-Biol. Interact. 2017, 276, 155–159.
- Balestri, F.; Poli, G.; Pineschi, C.; Moschini, R.; Cappiello, M.; Mura, U.; Tuccinardi, T.; Del Corso, A. Aldose Reductase Differential Inhibitors in Green Tea. Biomolecules 2020, 10, 1003.
- Cappiello, M.; Balestri, F.; Moschini, R.; Mura, U.; Del-Corso, A. Intra-site differential inhibition of multi-specific enzymes. J. Enzym. Inhib. Med. Chem. 2020, 35, 840–846.
- Balestri, F.; Cappiello, M.; Moschini, R.; Rotondo, R.; Buggiani, I.; Pelosi, P.; Mura, U.; Del-Corso, A. l-Idose: An attractive substrate alternative to d-glucose for measuring aldose reductase activity. Biochem. Biophys. Res. Commun. 2015, 456, 891–895.
- Misuri, L.; Cappiello, M.; Balestri, F.; Moschini, R.; Barracco, V.; Mura, U.; Del-Corso, A. The use of dimethylsulfoxide as a solvent in enzyme inhibition studies: The case of aldose reductase. J. Enzym. Inhib. Med. Chem. 2017, 32, 1152–1158.
- Balestri, F.; Quattrini, L.; Coviello, V.; Sartini, S.; Da Settimo, F.; Cappiello, M.; Moschini, R.; Del Corso, A.; Mura, U.; La Motta, C. Acid Derivatives of Pyrazolopyrimidine as Aldose Reductase Differential Inhibitors. Cell Chem. Biol. 2018, 25, 1414–1418.

