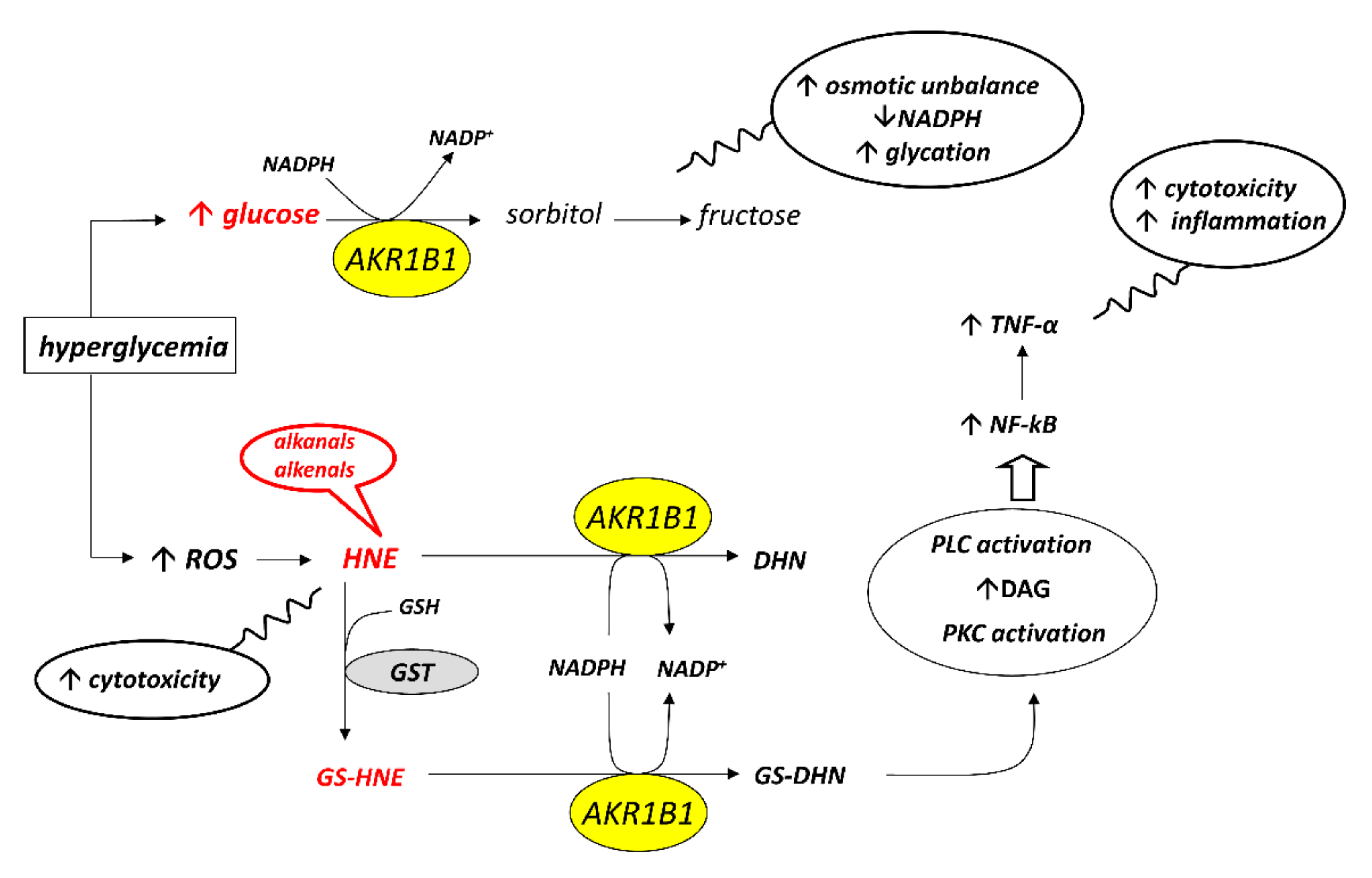
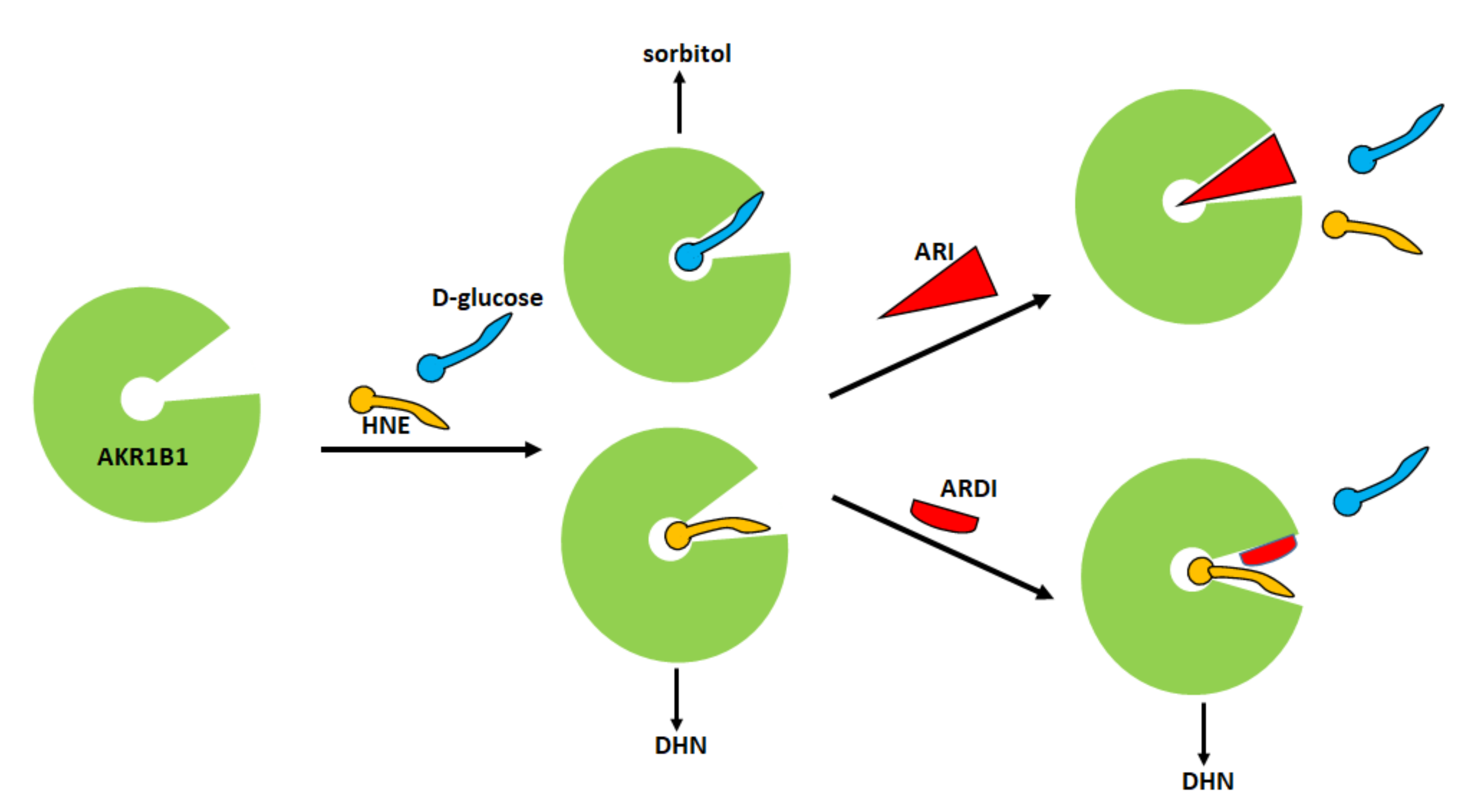
Starting from this first experimental evidence, a subsequent screening has been undertaken on a series of compounds previously developed as classical inhibitors of AKR1B1, in order to test the possibility that some of them could act as ARDIs. The possibility that different substrates bind differently to specific parts of the enzyme
[56][47] and the fact that the hemiacetal form of long-chain aldoses can modulate the activity of AKR1B1
[26][45] has raised the more general question of which substrate should be used in screening studies of AKR1B1 inhibitors. Most inhibition studies use
d,l-glyceraldehyde, a triose, which, in the light of the above considerations, cannot be considered the model substrate of the enzyme. Thus, in studies in which molecules for the inhibition of the reduction of
d-glucose by AKR1B1 are searched for,
d-glucose should be used as the appropriate substrate. However,
d-glucose, due to the very high K
M of AKR1B1 for this sugar, is a very poor substrate for the enzyme. Therefore, either large amounts of enzyme or extremely high glucose concentrations are required to have acceptable rate measurements. To circumvent the methodological limitations posed by the use of
d-glucose,
l-idose, the C-5 epimer of
d-glucose, which mimics the structure of
d-glucose, has been proposed as a good substrate alternative to
d-glucose
[28][49] and should be routinely used for searching for ARDIs. Furthermore, the evidence that dimethylsulfoxide (DMSO)—the solvent most frequently used to solubilize compounds during AKR1B1 inhibition studies—affects
l-idose and HNE reduction through different inhibition mechanisms
[58][50] highlighted the need to strictly control the solvent concentration especially in studies looking for ARDIs.
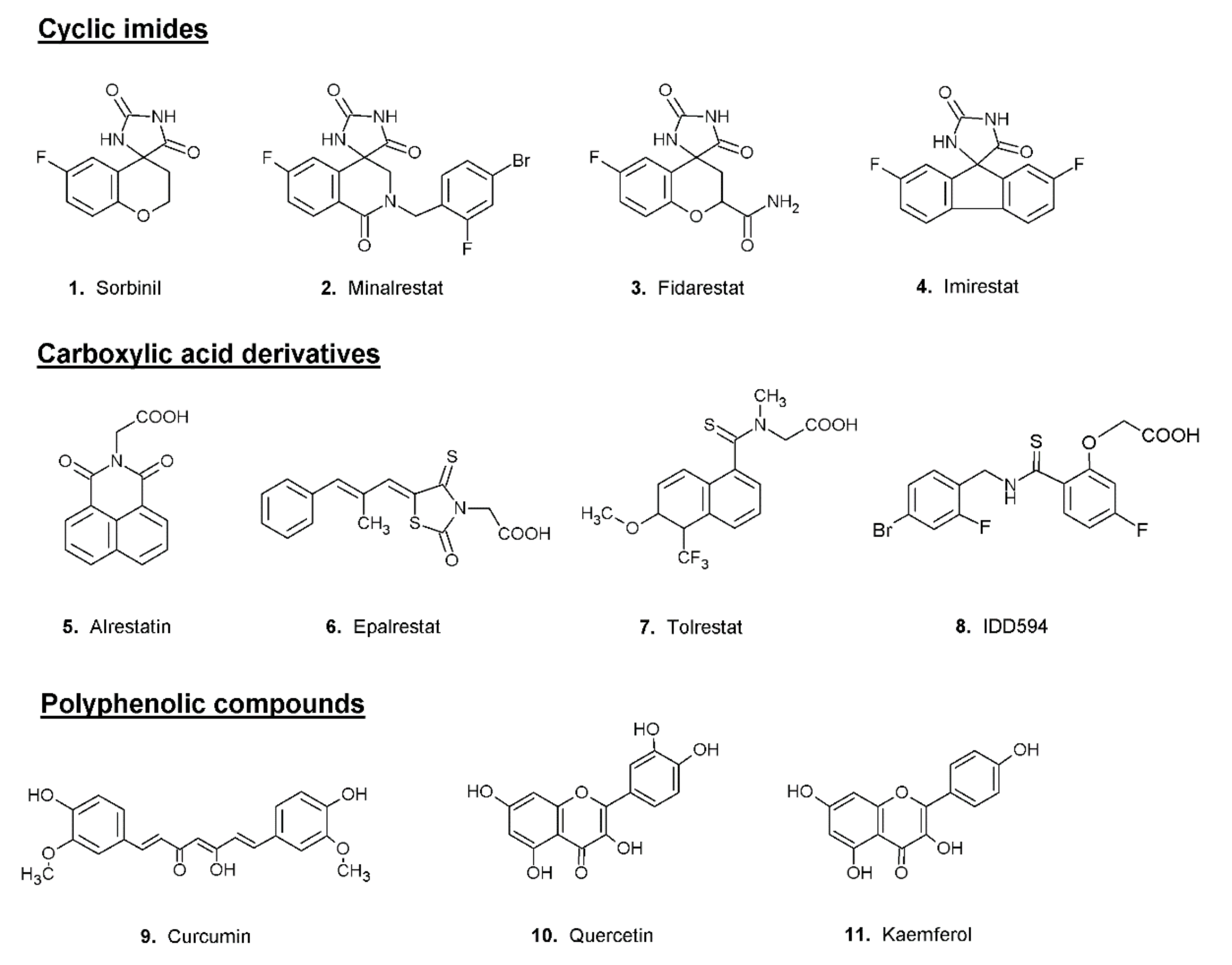
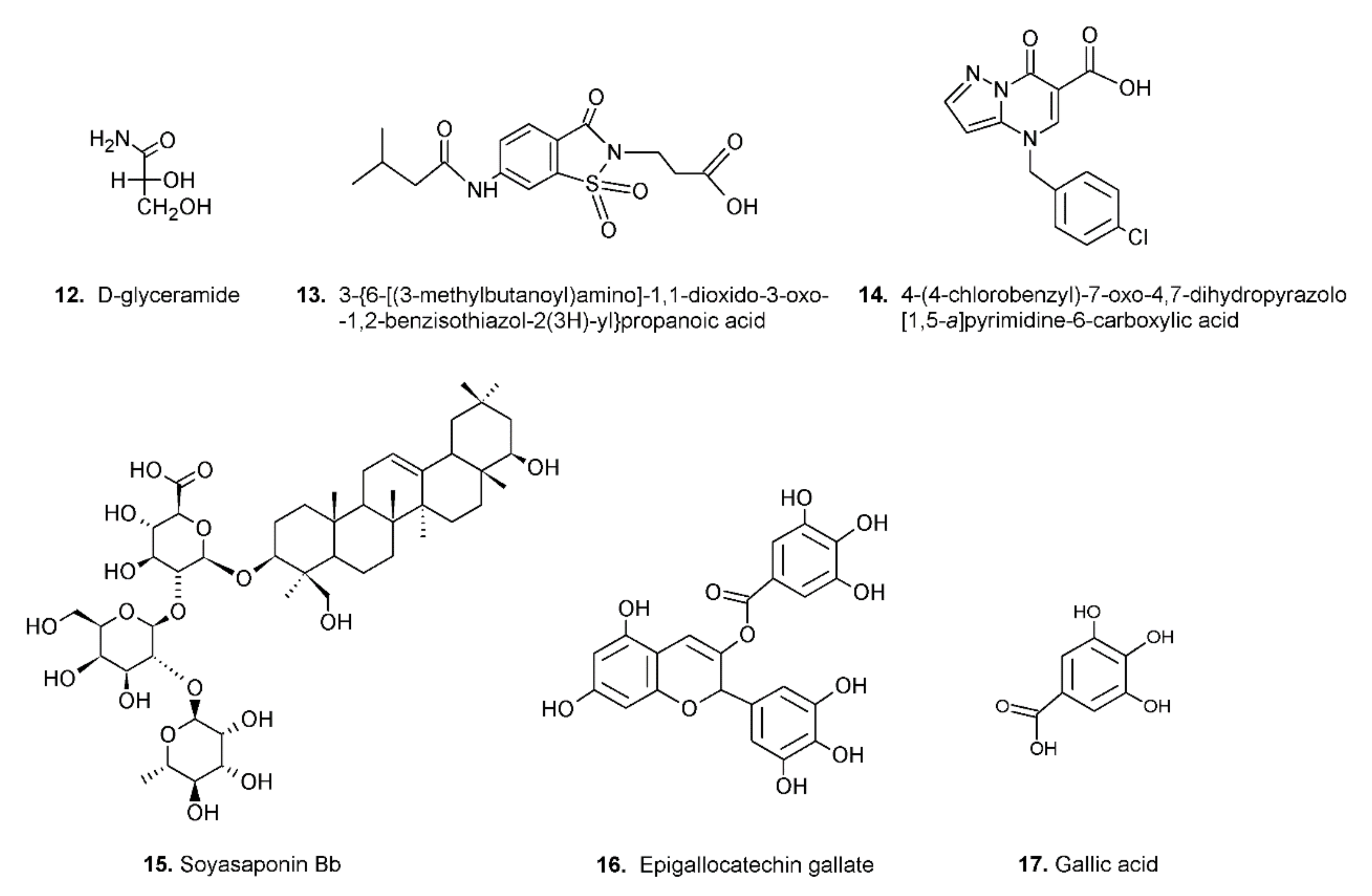
The search for ARDIs among some previously described AKR1B1 inhibitors evidenced that classical potent AKR1B1 inhibitors, such as Epalrestat and Sorbinil, showed no ability to differentially inhibit AKR1B1. At the same time, it has been shown that other molecules are able to modulate the activity of the enzyme depending on the specific substrate (
l-idose, HNE or GS-HNE adduct). In particular, when screening molecules of a class of acid derivatives of pyrazolo[1,5-
a] pyrimidine compounds (compound
14 of
Figure 4), characterized by the presence of a pyrazolo[1,5-
a] pyrimidine core and a carboxylic moiety, it has been reported that some compounds in this class can preferentially inhibit the reduction of sugars and GS-HNE adduct with respect to HNE
[59][51]. Explaining the structural features required for a molecule capable of inhibiting a certain mechanism of action while leaving an alternative pathway unaffected is not easy. The extensive structural information available for AKR1B1 is less helpful than one would expect, since it is lacking the most relevant information concerning the reciprocal influence between the substrate and the inhibitor both positioned on the enzyme. At the level of pure speculation, one might hypothesize that compounds able to preferentially inhibit the reduction of HNE over the reduction of glucose are able to somehow block the opening of the specificity pocket that allocates the hydrophobic tail of the HNE. This hypothesis might not be valid in the case of molecules which preferentially inhibit the reduction of glucose. In this case, the picture is further complicated by the presence of the hemiacetal form of glucose that influences the activity of AKR1B1.