 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Magdalena Druszczyńska | -- | 4859 | 2022-03-31 10:26:02 | | | |
| 2 | Nora Tang | Meta information modification | 4859 | 2022-04-01 08:09:03 | | |
Video Upload Options
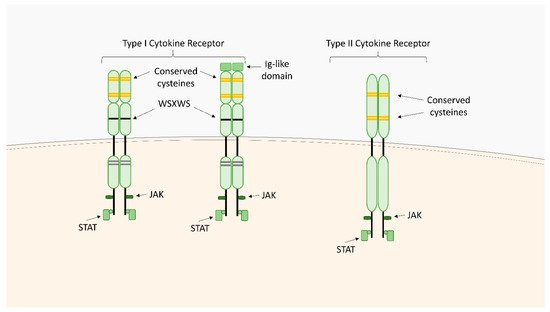
Cytokine receptors are membrane-bound or soluble glycoproteins that serve as cytokine docking sites and inductors of a signaling cascade inside the cells. They are involved in the initiation of intracellular signaling that regulates a diverse range of biological functions including metabolism control, neural stem cell activation, inflammatory responses as well as blood cell and immune cell development and growth. The classification of cytokine receptor families is based on the structural homology of the extracellular cytokine binding domains and common intracellular signaling mechanisms. The main families include type I cytokine receptors, type II cytokine receptors, chemokine receptors, the tumor necrosis factor (TNF) receptor family, the transforming growth factor (TGF)-β receptor family, the immunoglobulin (Ig) superfamily, and the interleukin (IL)-17 receptor family.
1. Type I Cytokine Receptors
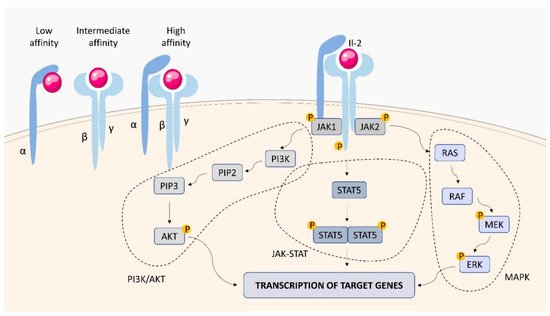
1.1. IL-2 Receptor (IL-2R)
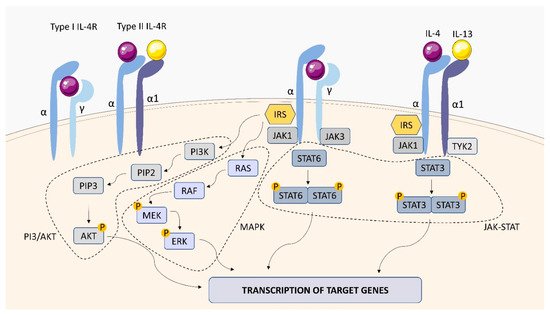
1.2. IL-4 Receptor (IL-4R)
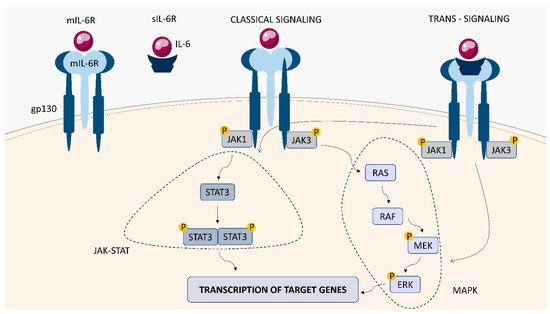
1.3. IL-6 Receptor (IL-6R)
1.4. IL-12 Receptor (IL-12R)
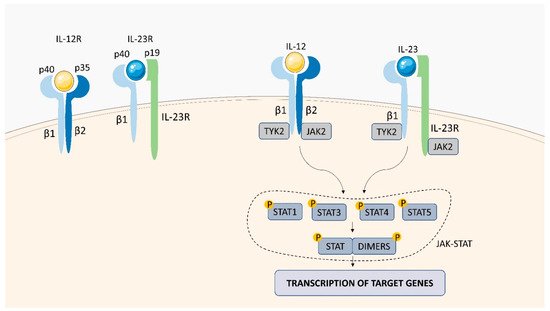
1.5. IL-23 Receptor (IL-23R)
2. Type II Cytokine Receptors
2.1. Interferon (IFN)-Gamma (γ) Receptor (IFN-γR)
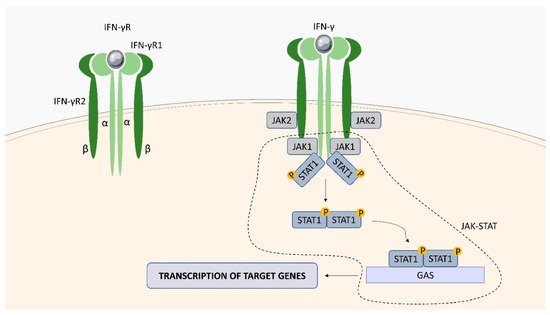
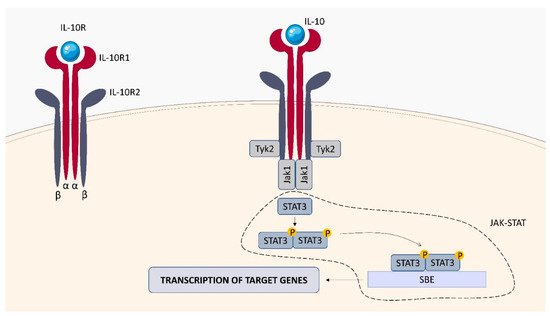
2.2. IL-10 Receptor (IL-10R)
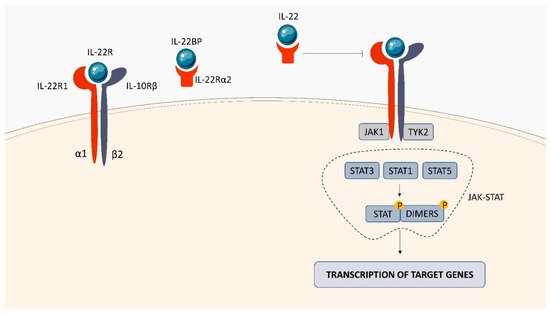
2.3. IL-22 Receptor (IL-22R)
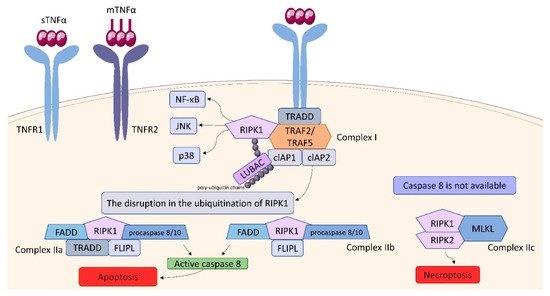
3. Tumor Necrosis Factor (TNF) Receptor (TNFR) Superfamily
References
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984.
- Fujii, H. Mechanisms of Signal Transduction from Receptors of Type I and Type II Cytokines. J. Immunotoxicol. 2007, 4, 69–76.
- Esen, İ. The Structure and Signaling Mechanisms of Type 1 Cytokine Receptors: A Brief Overview Tip 1 Sitokin Reseptörlerinin Yapısı ve Sinyal Mekanizmaları: Kısa Bir Genel Bakış. Turk. J. Immunol. 2015, 3, 121–124.
- Malek, T.R.; Castro, I. Interleukin-2 receptor signaling: At the interface between tolerance and immunity. Immunity 2010, 33, 153–165.
- Damoiseaux, J. The IL-2 - IL-2 receptor pathway in health and disease: The role of the soluble IL-2 receptor. Clin. Immunol. 2020, 218, 108515.
- Lawn, S.D.; Rudolph, D.; Ackah, A.; Coulibaly, D.; Wiktor, S.; Lal, R.B. Lack of induction of interleukin-2-receptor-α in patients with tuberculosis and human immunodeficiency virus co-infection: Implications for pathogenesis. Trans. R. Soc. Trop. Med. Hyg. 2001, 95, 449–452.
- Ryu, Y.J.; Ryu, K.H.; Kim, S.H.; Lee, J.S.; Cheon, S.H.; Seoh, J.Y. Soluble IL-2R, IFN-γ and neopterin as immunologic markers in patients with tuberculosis. Tuberc. Respir. Dis. 2002, 53, 294–308.
- Lee, S.-I.; Jin, H.-S.; Park, S. Association of genetic polymorphism of IL-2 receptor subunit and tuberculosis case . Biomedical Science Letters. Korean Soc. Biomed. Lab. Sci. 2018, 24, 94–101.
- Chendi, B.H.; Tveitenc, H.; Snyders, C.I.; Tonbya, K.; Jenumd, S.; Dam Nielsene, S.; Hove-Skovsgaarde, M.; Walzl, G.; Chegoub, N.N.; Dyrhol-Riise, A.M. CCL1 and IL-2Ra differentiate Tuberculosis disease from latent infection Irrespective of HIV infection in low TB burden countries. J. Infect. 2021, 83, 433–443.
- Cheung, L.S. Therapeutic Potential of an IL-2 Fusion Toxin in Tuberculosis and Melanoma. Doctoral Dissertations, Johns Hopkins University, Baltimore, Maryland, 2018. Available online: http://jhir.library.jhu.edu/handle/1774.2/61021 (accessed on 28 December 2021).
- Junttila, I.S. Tuning the Cytokine Responses: An Update on Interleukin (IL)-4 and IL-13 Receptor Complexes. Front. Immunol. 2018, 9, 888.
- Nelms, K.; Keegan, A.D.; Zamorano, J.; Ryan, J.J.; Paul, W.E. The IL-4 receptor: Signaling mechanisms and biologic functions. Annu. Rev. Immunol. 1999, 17, 701–738.
- Andrews, A.-L.; Holloway, J.W.; Holgate, S.T.; Davies, D.E. IL-4 receptor alpha is an important modulator of IL-4 and IL-13 receptor binding: Implications for the development of therapeutic targets. J. Immunol. 2006, 176, 7456–7461.
- Hölscher, C.; Heitmann, L.; Owusu-Dabo, E.; Horstmann, R.D.; Meyer, C.G.; Ehlers, S.; Thye, T. A mutation in IL4RA is associated with the degree of pathology in human TB patients. Mediat. Inflamm. 2016, 2016, 4245028.
- Heitmann, L.; Abad Dar, M.; Schreiber, T.; Erdmann, H.; Behrends, J.; Mckenzie, A.N.; Brombacher, F.; Ehlers, S.; Hölscher, C. The IL-13/IL-4Rα axis is involved in tuberculosis-associated pathology. J. Pathol. 2014, 234, 338–350.
- Parihar, S.P.; Ozturk, M.; Höft, M.A.; Chia, J.E.; Guler, R.; Keeton, R.; van Rensburg, I.C.; Loxton, A.G.; Brombacher, F. IL-4-responsive B cells are detrimental during chronic tuberculosis infection in mice. Front. Immunol. 2021, 12, 611673.
- Mihara, M.; Hashizume, M.; Yoshida, H.; Suzuki, M.; Shiina, M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin. Sci. 2012, 122, 143–159.
- Simpson, R.J.; Hammacher, A.; Smith, D.K.; Matthews, J.M.; Ward, L.D. Interleukin-6: Structure-function relationships. Protein Sci. 1997, 6, 929–955.
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295.
- Boni, F.G.; Hamdi, I.; Koundi, L.M.; Shrestha, K.; Xie, J. Cytokine storm in tuberculosis and IL-6 involvement. Infect. Genet. Evol. 2021, 97, 105166.
- Domingo-Gonzalez, R.; Prince, O.; Cooper, A.; Khader, S.A. Cytokines and chemokines in Mycobacterium tuberculosis infection. In Tuberculosis and the Tubercle Bacillus, 2nd ed.; William, R.J., Jr., Helen, M., Valerie, M., Ian, M.O., Eds.; ASM Press: Washington, DC, USA, 2017; pp. 33–72.
- Rantala, A.; Lajunen, T.; Juvonen, R.; Silvennoinen-Kassinen, S.; Peitso, A.; Vainio, O.; Saikku, P.; Leinonen, M. Association of IL-6 and IL-6R gene polymorphisms with susceptibility to respiratory tract infections in young Finnish men. Hum. Immunol. 2011, 72, 63–68.
- Ritter, K.; Sodenkamp, J.C.; Hölscher, A.; Behrends, J.; Hölscher, C. IL-6 Is Not Absolutely Essential for the Development of a TH17 Immune Response after an Aerosol Infection with Mycobacterium tuberculosis H37RV. Cells 2021, 10, 9.
- Delgobo, M.; Mendes, D.A.; Kozlova, E.; Rocha, E.L.; Rodrigues-Luiz, G.F.; Mascarin, L.; Dias, G.; Patrício, D.O.; Dierckx, T.; Bicca, M.A.; et al. An evolutionary recent IFN/IL-6/CEBP axis is linked to monocyte expansion and tuberculosis severity in humans. eLife 2019, 8, e47013.
- Hamza, T.; Barnett, J.B.; Li, B. Interleukin 12 a key immunoregulatory cytokine in infection applications. Int. J. Mol. Sci. 2010, 11, 789–806.
- Sinlgaglia, F.; Ambrosio, D.D.; Panina-Bordignon, P.; Rogse, L. Regulation of the IL-12/IL-12R axis: A critical step in T-helper cell differentiation and effector function. Immunol. Rev. 1999, 170, 65–72.
- Wu, C.Y.; Warrier, R.R.; Carvajal, D.M.; Chua, A.O.; Minetti, L.J.; Chizzonite, R.; Mongini, P.K.A.; Stern, A.S.; Gubler, U.; Presky, D.H.; et al. Biological function and distribution of human interleukin-12 receptor β chain. Eur. J. Immunol. 1996, 26, 345–350.
- Zhang, M.; Gong, J.; Presky, D.H.; Xue, W.; Barnes, P.F. Expression of the IL-12 Receptor β1 and β2 Subunits in Human Tuberculosis. J. Immunol. 1999, 162, 2441–2447.
- Bloch, Y.; Bouchareychas, L.; Merceron, R.; Składanowska, K.; Van den Bossche, L.; Detry, S.; Govindarajan, S.; Elewaut, D.; Haerynck, F.; Dullaers, M.; et al. Structural activation of pro-inflammatory human cytokine IL-23 by cognate IL-23 receptor enables recruitment of the shared receptor IL-12Rβ1. Immunity 2018, 48, 45.
- Tang, C.; Chen, S.; Qian, H.; Huang, W. Interleukin-23: As a drug target for autoimmune inflammatory diseases. Immunology 2012, 135, 112–124.
- Pastor-Fernández, G.; Mariblanca, I.R.; Navarro, M.N. Decoding IL-23 Signaling Cascade for New Therapeutic Opportunities. Cells 2020, 9, 2044.
- Shen, H.; Chen, Z.W. The crucial roles of Th17-related cytokines/signal pathways in M. tuberculosis infection. Cell. Mol. Immunol. 2018, 15, 216–225.
- Khader, S.A.; Bell, G.K.; Pearl, J.E.; Fountain, J.J.; Rangel-Moreno, J.; Cilley, G.E.; Shen, F.; Eaton, S.M.; Gaffen, S.L.; Swain, S.L.; et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat. Immunol. 2007, 84, 369–377.
- Shen, H.; Wang, Y.; Chen, C.Y.; Frencher, J.; Huang, D.; Yang, E.; Ryan-Payseur, B.; Chen, Z.W. Th17-related cytokines contribute to recall-like expansion/effector function of HMBPP-specific Vγ2Vδ2 T cells after Mycobacterium tuberculosis infection or vaccination. Eur. J. Immunol. 2015, 45, 442–451.
- McKenzie, B.S.; Kastelein, R.A.; Cua, D.J. Understanding the IL-23–IL-17 immune pathway. Trends Immunol. 2006, 27, 17–23.
- Shen, H.; Gu, J.; Xiao, H.; Liang, S.; Yang, E.; Yang, R.; Huang, D.; Chen, C.; Wang, F.; Shen, L.; et al. Selective Destruction of Interleukin 23–Induced Expansion of a Major Antigen–Specific γδ T-Cell Subset in Patients With Tuberculosis. J. Infect. Dis. 2017, 215, 420.
- Renauld, J.C. Class II cytokine receptors and their ligands: Key antiviral and inflammatory modulators. Nat. Rev. Immunol. 2003, 38, 667–676.
- Ivashkiv, L.B. IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545.
- Tau, G.; Rothman, P. Biologic functions of the IFN-gamma receptors. Allergy 1999, 54, 1233–1251.
- Kak, G.; Tiwari, B.K.; Singh, Y.; Natarajan, K. Regulation of Interferon-γreceptor (IFN-γR) expression in macrophages during Mycobacterium tuberculosis infection. Biomol. Concepts 2020, 11, 76–85.
- Cooke, G.S.; Campbell, S.J.; Sillah, J.; Gustafson, P.; Bah, B.; Sirugo, G.; Bennett, S.; McAdam, K.P.W.J.; Sow, O.; Lienhardt, C.; et al. Polymorphism within the Interferon-γ/Receptor Complex Is Associated with Pulmonary Tuberculosis. Am. J. Respir. Crit. Care Med. 2006, 174, 339–343.
- Singhal, A.; Jaiswal, A.; Arora, V.K.; Prasad, H.K. Modulation of gamma interferon receptor 1 by Mycobacterium tuberculosis: A potential immune response evasive mechanism. Infect. Immun. 2007, 75, 2500–2510.
- Gupta, D.; Sharma, S.; Singhal, J.; Satsangi, A.T.; Antony, C.; Natarajan, K. Suppression of TLR2-induced IL-12, reactive oxygen species, and inducible nitric oxide synthase expression by Mycobacterium tuberculosis antigens expressed inside macrophages during the course of infection. J. Immunol. 2010, 184, 5444–5455.
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777.
- Iyer, S.S.; Cheng, G. Role of Interleukin 10 Transcriptional Regulation in Inflammation and Autoimmune Disease. Crit. Rev. Immunol. 2012, 32, 23.
- Shouval, D.S.; Ouahed, J.; Biswas, A.; Goettel, J.A.; Horwitz, B.H.; Klein, C.; Muise, A.M.; Snapper, S.B. Interleukin 10 Receptor Signaling: Master Regulator of Intestinal Mucosal Homeostasis in Mice and Humans. Adv. Immunol. 2014, 122, 177.
- Murray, P.J. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc. Natl. Acad. Sci. USA 2005, 102, 8686–8691.
- Turner, J.; Gonzalez-Juarrero, M.; Ellis, D.L.; Basaraba, R.J.; Kipnis, A.; Orme, I.M.; Cooper, A.M. In vivo IL-10 production reactivates chronic pulmonary tuberculosis in C57BL/6 mice. J. Immunol. 2002, 169, 6343–6351.
- Redford, P.S.; Boonstra, A.; Read, S.; Pitt, J.; Graham, C.; Stavropoulos, E.; Bancroft, G.J.; O’Garra, A. Enhanced protection to Mycobacterium tuberculosis infection in IL-10-deficient mice is accompanied by early and enhanced Th1 responses in the lung. Eur. J. Immunol. 2010, 40, 2200–2210.
- Beamer, G.L.; Flaherty, D.K.; Assogba, B.D.; Stromberg, P.; Gonzalez-Juarrero, M.; de Waal Malefyt, R.; Vesosky, B.; Turner, J. Interleukin-10 promotes Mycobacterium tuberculosis disease progression in CBA/J mice. J. Immunol. 2008, 181, 5545–5550.
- Cyktor, J.C.; Carruthers, B.; Kominsky, R.A.; Beamer, G.L.; Stromberg, P.; Turner, J. IL-10 inhibits mature fibrotic granuloma formation during Mycobacterium tuberculosis infection. J. Immunol. 2013, 190, 2778–2790.
- Pitt, J.M.; Stavropoulos, E.; Redford, P.S.; Beebe, A.M.; Bancroft, G.J.; Young, D.B.; O’Garra, A. Blockade of IL-10 signaling during BCG vaccination enhances and sustains Th1, Th17, and innate lymphoid IFN-γ and IL-17 responses and increases protection to Mycobacterium tuberculosis infection. J. Immunol. 2012, 189, 4079.
- Dwivedi, V.; Gautam, S.; Headley, C.A.; Piergallini, T.; Torrelles, J.B.; Turner, J. IL-10 receptor blockade delivered simultaneous with BCG vaccination sustains long term protection against Mycobacterium tuberculosis infection in mice. BioRxiv. 2021, 458995.
- Zenewicz, L.A. IL-22: There Is a Gap in Our Knowledge. ImmunoHorizons 2018, 2, 198–207.
- Dudakov, J.A.; Hanash, A.M.; Van Den Brink, M.R.M. Interleukin-22: Immunobiology and pathology. Annu. Rev. Immunol. 2015, 33, 747–785.
- Parks, O.B.; Pociask, D.A.; Hodzic, Z.; Kolls, J.K.; Good, M. Interleukin-22 signaling in the regulation of intestinal health and disease. Front. Cell Dev. Biol. 2016, 3, 85.
- Arshad, T.; Mansur, F.; Palek, R.; Manzoor, S.; Liska, V. A Double Edged Sword Role of Interleukin-22 in Wound Healing and Tissue Regeneration. Front. Immunol. 2020, 11, 2148.
- Aujla, S.J.; Chan, Y.R.; Zheng, M.; Fei, M.; Askew, D.J.; Pociask, D.A.; Reinhart, T.A.; McAllister, F.; Edeal, J.; Gaus, K.; et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 2008, 14, 275.
- Zeng, G.; Chen, C.Y.; Huang, D.; Yao, S.; Wang, R.C.; Chen, Z.W. Membrane-bound IL-22 after de novo production in tuberculosis and anti-M.tuberculosis effector function of IL-22+CD4+ T cells. J. Immunol. 2011, 187, 190.
- Ronacher, K.; Sinha, R.; Cestari, M. IL-22: An Underestimated Player in Natural Resistance to Tuberculosis? Front. Immunol. 2018, 9, 2209.
- Whittington, H.A.; Armstrong, L.; Uppington, K.M.; Millar, A.B. Interleukin-22: A potential immunomodulatory molecule in the lung. Am. J. Respir. Cell Mol. Biol. 2004, 31, 220–226.
- Treerat, P.; Prince, O.; Cruz-Lagunas, A.; Muñoz-Torrico, M.; Salazar-Lezama, M.A.; Selman, M.; Fallert-Junecko, B.; Reinhardt, T.A.; Alcorn, J.F.; Kaushal, D.; et al. Novel role for IL-22 in protection during chronic Mycobacterium tuberculosis HN878 infection. Mucosal Immunol. 2017, 10, 1069–1081.
- Chu, W.M. Tumor necrosis factor. Cancer Lett. 2013, 328, 222.
- Segueni, N.; Benmerzoug, S.; Rose, S.; Gauthier, A.; Bourigault, M.L.; Reverchon, F.; Philippeau, A.; Erard, F.; Le Bert, M.; Bouscayrol, H.; et al. Innate myeloid cell TNFR1 mediates first line defence against primary Mycobacterium tuberculosis infection. Sci. Rep. 2016, 6, 22454.
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF family of ligands and receptors: Communication modules in the immune system and beyond. Physiol. Rev. 2019, 99, 115–160.
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M. Tumour necrosis factor signalling in health and disease. F1000Res 2019, 8, 111.
- Idriss, H.T.; Naismith, J.H. TNF alpha and the TNF receptor superfamily: Structure-function relationship(s). Microsc. Res. Tech. 2000, 50, 184–195.
- Tartaglia, L.A.; Pennica, D.; GoeddelS, D.V. Ligand Passing: The 75-kDa Tumor Necrosis Factor (TNF) Receptor Recruits TNF for Signaling by the 55-kDa TNF’ Receptor*. J. Biol. Chem. 1993, 268, 18542–18548.
- Keeton, R.; du Toit, J.P.; Hsu, N.J.; Dube, F.; Jacobs, M. Immune control of Mycobacterium tuberculosis is dependent on both soluble TNFRp55 and soluble TNFRp75. Immunology 2021, 164, 524–540.
- Hijdra, D.; Vorselaars, A.D.; Grutters, J.C.; Claessen, A.M.; Rijkers, G.T. Differential expression of TNFR1 (CD120a) and TNFR2 (CD120b) on subpopulations of human monocytes. J. Inflamm. 2012, 9, 38.
- Gough, P.; Myles, I.A. Tumor Necrosis Factor Receptors: Pleiotropic Signaling Complexes and Their Differential Effects. Front. Immunol. 2020, 11, 585880.
- Lavrik, I.; Golks, A.; Krammer, P.H. Death receptor signaling. J. Cell Sci. 2005, 118, 265–267.
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190.

