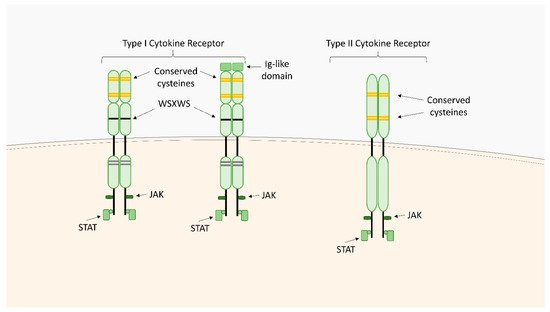
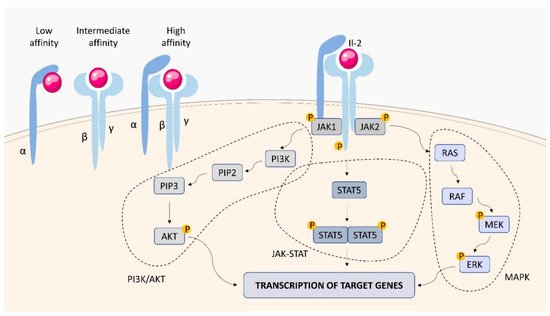
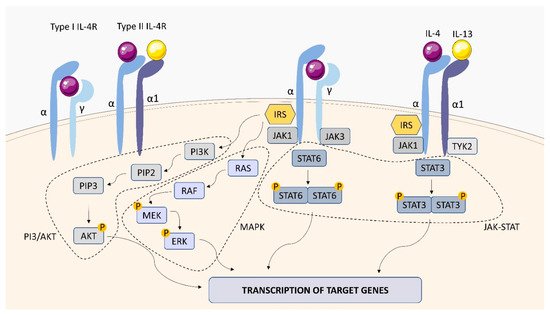
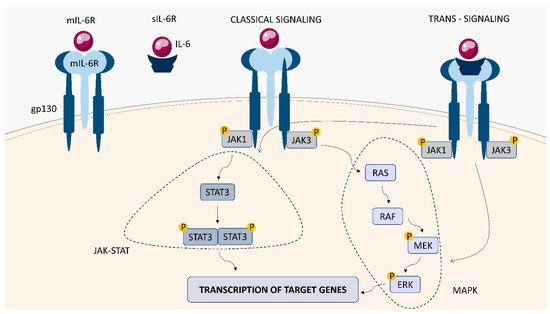
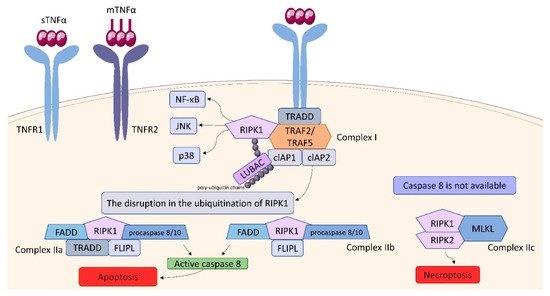
Within the TNFSF/TNFRSF superfamily consisting of 19 ligands and 29 receptors, TNF-α and its two main receptors: TNFR1 (TNFRp55/CD120a) and TNFR2 (TNFRp75/CD120b) are among the best characterized members. Soluble TNF-α (sTNF-α) preferentially binds to TNFR1, while membrane bound TNF-α (mTNF-α) is attached to TNFR2
[63][64][65][66][67][103,104,105,106,107]. Both receptors are single transmembrane glycoproteins
[68][108]. Similarly to TNF-α, both TNFR1 and TNFR2, are synthesized in a membrane-bound form, which can be cleaved by metalloproteases to release soluble receptors (sTNFR)
[69][109]. TNFR1 is expressed on nearly all nucleated cells
[70][110]. As a death domain (DD) is present in the structure of TNFR1, this receptor belongs to cell sensors termed death receptors (DR)
[71][111]. Two types of the DR signaling complex have been distinguished: the first group comprises the death-inducing signaling complexes (DISCs) and the second one, including TNFR1, transduces both apoptotic and survival signals
[72][112]. TNFR1 is constitutively expressed on most cell types and undergoes activation not only by sTNF-α but also by mTNF-α
[71][111]. TNFR1 stimulation is associated with the formation of two signaling complexes: Complex I, which leads to cytokine signaling and cell survival via activation of NF-kB, JNK, and p38 pathways, Complex II (in different variants) leading to apoptotic or necrotic cell death
[71][73][111,113] (
Figure 910). TNF binding toTNFR1 entails trimerization of the receptor and formation of a core signaling complex within the cytoplasmic tail of TNFR1. Trimerization enables the recruitment of TRADD (TNFR1-associated death domain) through DD. Acting as a scaffold, TRADD recruits the next elements of the forming complex: receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and TNF receptor-associated factor (TRAF) 2, or TRAF5, whereby TRAF2 is responsible for recruiting other elements: cellular inhibitor of apoptosis protein (cIAP) 1 and cIAP2. Thus, the core signaling complex formed on the membrane consists of TRADD, TRAF2 (or TRAF5), RIPK1, and cIAP1/2. There are two factors determining whether it will be a platform for developing Complex I or Complex II: ubiquitination of RIPK1 and the availability of caspase molecules. As a result of the activity of TRAF and cIAP1/2, the ubiquitin chains are added to RIPK1 acting as scaffolds for other signaling factors, including K63, K11, K48 poly-ubiquitin chains and the linear ubiquitin chain assembly complex (LUBAC). The final stage in the formation of Complex I is the attachment of an M1 polyubiquitin chain to RIPK1 by LUBAC. Fully assembled Complex I activates NF-kB, JNK, and p38 pathways via recruitment of the inhibitor of the IkB kinase (IKK) complex and the TGFβ-activated kinase 1 (TAK1)-dependent mechanism, as it was described in detail by Gough and Myles
[71][111]. The disruption in the ubiquitination of RIPK1 resulting in its release into cytosol leads to the formation of Complex II, which can be assembled in two variants: IIa and IIb. The attachment of TRADD, Fas-associated death domain (FADD), FLICE-like inhibitory protein (FLIPL) and procaspase 8 or 10 to RIPK1 forms Complex IIa. Variant IIb includes the same components as Complex IIa, except it lacks TRADD. The assembly of these proteins allow converting procaspase 8/10 to the active form that triggers the cell death by apoptosis. If caspase 8 is not available, Complex IIc is formed. It involves RIPK1 and RIPK3 assembling in an amyloid-like structure to form the necrosome, which leads to cell lysis (necroptosis) via mixed-lineage kinase domain-like protein (MLKL) and phosphoinositides binding
[71][111]. It is suggested, that the effectiveness of complex II formation, caspase-8 activation, and the availability of FLIP in the cell cytosol, which prevents procaspase-8 activation at Complex II, determine whether a cell stays alive/active or dies. This model seems to be an attractive framework for making life-or-death decisions, however it requires more experimental proof
[64][104].