 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mariusz Mojzych | + 1858 word(s) | 1858 | 2022-03-07 02:47:52 | | | |
| 2 | Conner Chen | -6 word(s) | 1852 | 2022-03-22 06:54:34 | | |
Video Upload Options
Coumarin is an important six-membered aromatic heterocyclic pharmacophore, widely distributed in natural products and synthetic molecules. The versatile and unique features of coumarin nucleus, in combination with privileged sulfonamide moiety, have enhanced the broad spectrum of biological activities. The research and development of coumarin, sulfonamide-based pharmacology, and medicinal chemistry have become active topics, and attracted the attention of medicinal chemists, pharmacists, and synthetic chemists. Coumarin sulfonamide compounds and analogs as clinical drugs have been used to cure various diseases with high therapeutic potency, which have shown their enormous development value. The diversified and wide array of biological activities such as anticancer, antibacterial, anti-fungal, antioxidant and anti-viral, etc. were displayed by diversified coumarin sulfonamides.
1. Introduction
2. Coumarin Sulfonamides as Anti-Cancer Agents and Carbonic Anhydrase Inhibitors
2.1. Benzenesulfonamide-Based Coumarins as Carbonic Anhydrases II and IX Inhibitors
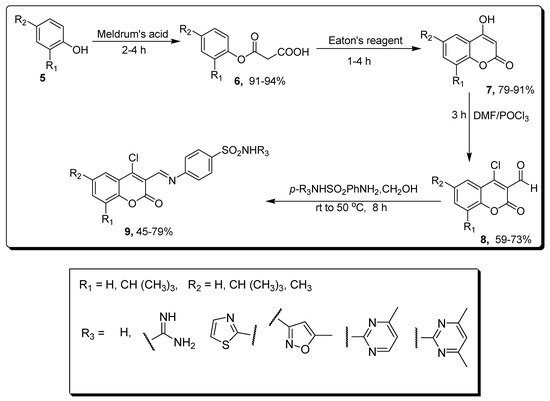
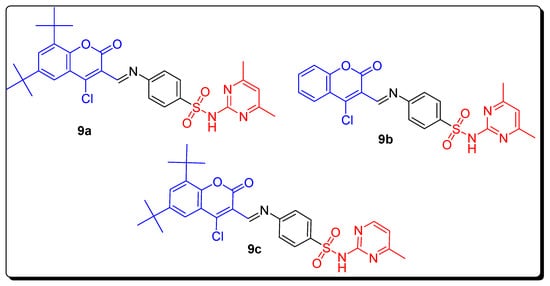
| Compound | MCF-7 µM | Compounds | hCAs II µM | hCAs IX µM |
|---|---|---|---|---|
| 9a | 0.0088 | 9b | - | 0.124 |
| Doxorubicin | 0.065 | 9c | 0.063 | - |
| Semaxanib | 0.0031 | AAZ | 0.016 | 0.028 |
| - | - | SA | 0.26 | 0.29 |
2.2. Thiazole-Sulfonamide Coumarin Hybrids as hCA I and hCA II Inhibitors
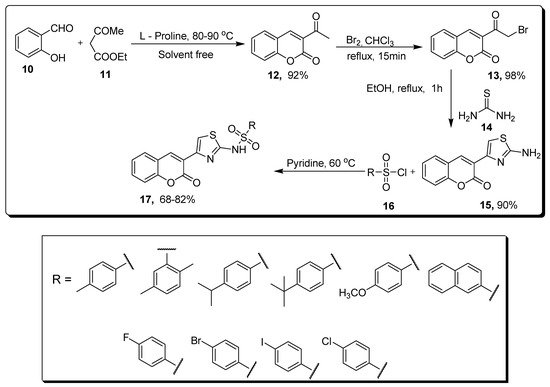
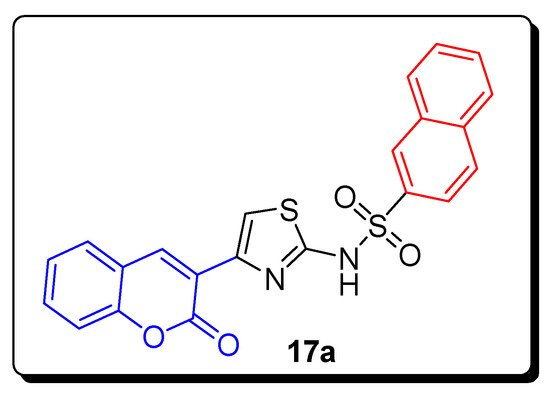
| Compound | hCA I IC50 (µM) |
hCA II IC50 (µM) |
|---|---|---|
| 17a | 5.63 | 8.48 |
2.3. Sulfonyl Ureido Coumarins Hybrids as Carbonic Anhydrase Inhibitors
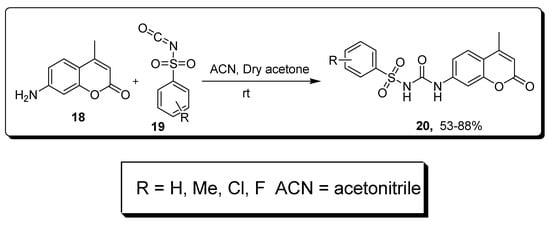
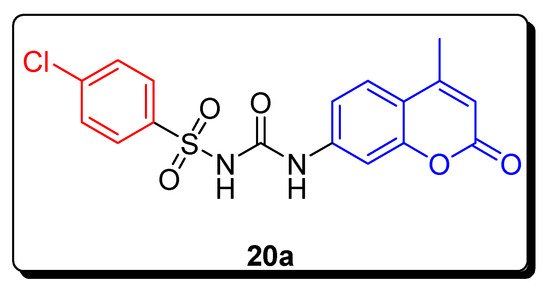
| Compound | hCA IX KI (nM) |
hCA XII KI (nM) |
|---|---|---|
| 20a | 20.2 | 6.0 |
| AAZ | 25.0 | 5.7 |
2.4. Benzene Sulfonamido-Coumarinyl Hydrazones Hybrids as CA Inhibitors
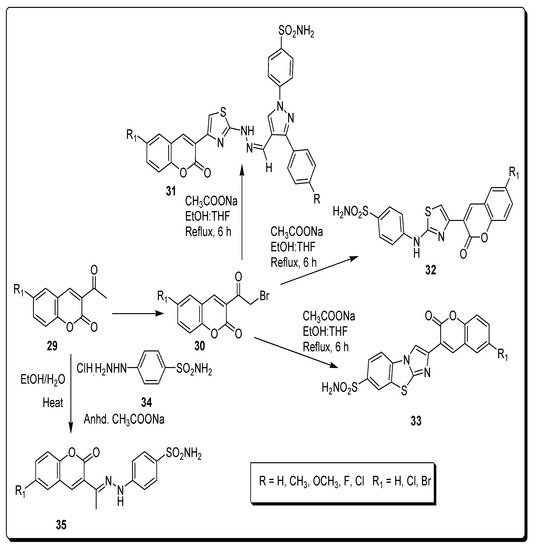
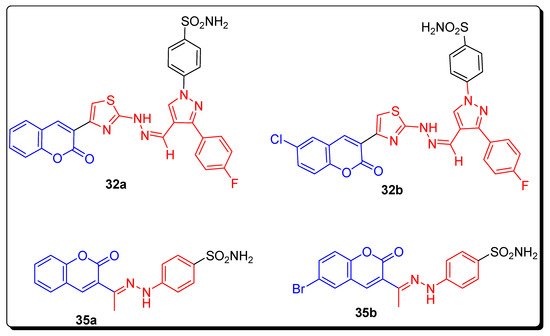
| Compound Number | hCA I KI (nM) |
hCA II KI (nM) |
hCA IX KI (nM) |
hCA XII KI (nM) |
|---|---|---|---|---|
| 32a | 263.49 | 21.20 | 2.28 | 0.54 |
| 32b | 349.63 | 17.46 | 2.54 | 0.54 |
| 35a | 220.13 | 13.23 | 58.61 | 4.4 |
| 35b | 21.95 | 1751.72 | 23.59 | 0.62 |
| AZA | 250.0 | 12.1 | 25.0 | 5.7 |
References
- De Souza, L.G.; Rennã, M.N.; Figueroa-Villar, J.D. Coumarins as cholinesterase inhibitors. A review. Chem. Biol. Interact. 2016, 254, 11–23.
- Peng, X.M.; Damu, L.V.; Zhou, C.H. Current developments of coumarin compounds in medicinal chemistry. Curr. Pharm. Des. 2013, 19, 3884–3930.
- Kostova, I. Synthetic and natural coumarins as antioxidants. Mini Rev. Med. Chem. 2006, 6, 365–374.
- Pereira, M.T.; Franco, P.D.; Vitorio, F.; Kümmerle, E.A. Coumarin compounds in medicinal chemistry: Some important examples from the last years. Curr. Top. Med. Chem. 2018, 18, 124–148.
- Murray, R.D.H. Coumarins. Nat. Prod. Rep. 1995, 12, 477–505.
- Pereira, T.M.; Vitório, F.; Amaral, R.C.; Zanoni, K.P.S.; Iha, N.Y.M.; Kümmerle, A.E. Microwave-assisted synthesis and photophysical studies of novel fluorescent N-acylhydrazone and semicarbazone-7-OH-coumarin dyes. New J. Chem. 2016, 40, 8846–8854.
- Symeonidis, T.; Chamilos, M.; Litina, D.J.H.; Kallitsakis, M.; Litinas, K.E. Synthesis of hydroxycoumarins and hydroxybenzo- or coumarins as lipid peroxidation inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 1139–1142.
- Sashidhara, K.V.; Kumar, A.; Kumar, J.M.; Sinha, S.S. Synthesis and in vitro evaluation of novel coumarinechalcone hybrids as potential anticancer agents. Bioorg. Med. Chem. Lett. 2010, 20, 7205–7721.
- Reddy, N.S.; Mallireddigari, M.R.; Cosenza, S.; Gumireddy, K.; Bell, S.C.; Reddy, E.P.; Reddy, M.R. Synthesis of new coumarin 3-(N-aryl) sulfonamides and their anticancer activity. Bioorg. Med. Chem. Lett. 2004, 14, 4093–4097.
- Nasr, S.T.; Bondock, M. Youns, Anticancer activity of new coumarin substituted hydrazide-hydrazone derivatives. Eur. J. Med. Chem. 2014, 76, 539–548.
- Thakur, A.; Singla, R.; Jaitak, V. Coumarins as anticancer agents: A review on synthetic strategies, mechanism of action and SAR studies. Eur. J. Med. Chem. 2015, 101, 476–495.
- Devji, T.; Reddy, C.; Woo, C.; Awale, S.; Kadota, S.; Carrico-Moniz, D. Pancreatic anticancer activity of a novel geranylgeranylatedcoumarin derivative. Bioorg. Med. Chem. Lett. 2011, 21, 5770–5773.
- Alshibl, H.M.; Al-Abdullah, E.S.; Haiba, M.E.; Alkahtani, H.M.; Awad, G.E.A.; Mahmoud, A.H.; Ibrahim, B.M.M.; Bari, A.; Villinger, A. Synthesis and Evaluation of New Coumarin Derivatives as Antioxidant, Antimicrobial, and Anti-Inflammatory Agents. Molecules 2020, 25, 3251.
- Melagraki, G.; Afantitis, A.; Igglessi-Markopoulou, O.; Detsi, A.; Koufaki, M.; Kontogiorgis, C.; Hadjipavlou-Litina, D.J. Synthesis and evaluation of the antioxidant and anti-inflammatory activity of novel coumarin-3-aminoamides and their alpha-lipoic acid adducts. Eur. J. Med. Chem. 2009, 44, 3020–3026.
- Fylaktakidou, K.C.; Hadjipavlou-Litina, D.J.; Litinas, K.E.; Nicolaides, D.N. Natural and synthetic coumarin derivatives with anti-inflammatory/antioxidant activities. Curr. Pharm. Des. 2004, 10, 3813–3833.
- Kontogiorgis, C.; Hadjipavlou-Litina, D. Biological evaluation of several coumarins derivatives designed as possible anti-inflammatory/antioxidant agents. J. Enzym. Inhib. Med. Chem. 2003, 18, 63–69.
- Rehman, S.U.; Chohan, Z.H.; Gulnaz, F.; Supuran, C.T. In-vitro antibacterial, antifungal and cytotoxic activities of some coumarins and their metal complexes. J. Enzym. Inhib. Med. Chem. 2005, 20, 333–340.
- Kalluraya, B.; Vishwanatha, P.; Isloor, A.M.; Rai, G.; Kotian, M. Synthesis and biological activity of 6-substituted-3- coumarins. Boll. Chim. Farm. 2000, 139, 263–266.
- Musiciki, B.; Periers, A.M.; Laurin, P.; Ferroud, D.; Benedetti, Y.; Lachaud, S.; Chatreaux, F.; Haesslein, J.L.; LLtis, A.; Pierre, C.; et al. Improved antibacterial activities of coumarin antibiotics bearing 5′,5′-dialkylnoviose: Biological activity of RU79115. Bioorg. Med. Chem. Lett. 2000, 10, 1695.
- De Souza, S.M.; Monache, F.D.; Smânia, A. Antibacterial activity of coumarins. Z. Nat. C 2005, 60, 693–700.
- Guerra, F.Q.S.; de Araújo, R.S.A.; de Sousa, J.P.; de Pereira, F.O.; Mendonça-Junior, F.J.B.; Barbosa-Filho, J.M.; de Oliveira Lima, E. Evaluation of antifungal activity and mode of action of new coumarin derivative, 7-hydroxy-6-nitro-2h-1-benzopyran-2-one, against Aspergillus spp. Evid. Based Complement. Altern. Med. 2015, 2015, 925096.
- Montagner, C.; de Souza, S.M.; Groposo, C.; DelleMonache, F.; Smânia, E.F.A.; Smânia, A., Jr. Antifungal activity of coumarins. Z. Nat. C J. Biosci. 2008, 63, 21–28.
- Sardari, S.; Mori, Y.; Horita, K.; Micetich, R.G.; Nishibe, S.; Daneshtalab, M. Synthesis and antifungal activity of coumarins and angular furanocoumarins. Bioorg. Med. Chem. 1999, 7, 1933–1940.
- Ghate, M.; Manohar, D.; Kulkarni, V.; Shobha, R.; Kattimani, S.Y. Synthesis of vanillin ethers from 4-(bromomethyl) coumarins as anti-inflammatory agents. Eur. J. Med. Chem. 2003, 38, 297–302.
- Christos, A.K.; Dimitra, J.H. Synthesis and anti-inflammatory activity of coumarin derivatives. J. Med. Chem. 2005, 48, 6400–6408.
- Li, H.; Yao, Y.; Li, L. Coumarins as potential antidiabetic agents. J. Pharm. Pharmacol. 2017, 69, 1253–1264.
- Campos-Toimil, M.; Orallo, F.; Santana, L.; Uriarte, E. Synthesis and vasorelaxant activity of new coumarin and furocoumarin derivatives. Bioorg. Med. Chem. Lett. 2002, 12, 783–786.
- Ghate, M.; Kusanur, R.A.; Kulkarni, M.V. Synthesis and in vivo analgesic and anti-inflammatory activity of some bi heterocyclic coumarin derivatives. Eur. J. Med. Chem. 2005, 40, 882–887.
- Kostova, I.; Raleva, S.; Genova, P.; Argirova, R. Structure-activity relationships of synthetic coumarins as hiv-1 inhibitors. Bioinorg. Chem. Appl. 2006, 2006, 68274.
- Ojala, T.; Remes, S.; Haansuu, P.; Vuorela, H.; Hiltunen, R.; Haatela, K.; Vuorela, P. Antimicrobial activity of some coumarin containing herbal plants growing in Finland. J. Ethnopharmacol. 2000, 73, 299–305.
- Abdelhafez, M.O.; Amin, M.K.; Batran, Z.R.; Maher, J.T.; Nada, A.S.; Sethumadhavan, S. Synthesis, anticoagulant and PIVKA-II induced by new 4-hydroxycoumarin derivatives. Bioorg. Med. Chem. 2010, 18, 3371–3378.
- Ahmad, R.; Asad, M.; Siddiqui, N.Z.; Kumar, A. Evaluation of antipyretic and antinociceptive potential of new heterocyclic derivatives of 3-formyl-4-hydroxycoumarin in rats. J. Pharm. Appl. Sci. 2013, 3, 253–259.
- Hassanpour, S.H.; Dehghani, M. Review of cancer from perspective of molecular. J. Cancer Res. Pract. 2017, 4, 127–129.
- Arruebo, M.; Vilaboa, N.; Sáez-Gutierrez, B.; Lambea, J.; Tres, A.; Valladares, M.; González-Fernández, Á. Assessment of the evolution of cancer treatment therapies. Cancers 2011, 3, 3279–3330.
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.J.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243.
- Irfan, A.; Rubab, L.; Rehman, U.M.; Anjum, R.; Ullah, S.; Marjana, M.; Qadeer, S.; Sana, S. Coumarin sulfonamide derivatives: An emerging class of therapeutic agents. Heterocycl. Commun. 2020, 26, 46–59.
- Supuran, C.T. Structure-based drug discovery of carbonic anhydrase inhibitors. J. Enzym. Inhib. Med. Chem. 2012, 27, 759–772.
- Wagner, J.; Avvaru, S.B.; Robbins, H.A.; Scozzafava, A.; Supuran, T.C.; McKenna, R. Coumarinyl-substituted sulfonamides strongly inhibit several human carbonic anhydrase isoforms: Solution and crystallographic investigations. Bioorg. Med. Chem. 2010, 18, 4873–4878.
- Supuran, C.T. Bacterial carbonic anhydrases as drug targets: Toward novel antibiotics. Front. Pharmacol. 2011, 2, 34.
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777.
- Supuran, C.T. Carbonic anhydrase inhibitors: An editorial. Expert Opin. Ther. Pat. 2013, 23, 677–679.
- Angeli, A.; Carta, F.; Supuran, C.T. Carbonic Anhydrases: Versatile and Useful Biocatalysts in Chemistry and Biochemistry. Catalysts 2020, 10, 1008.
- Capasso, C.; Supuran, C.T. An overview of the alpha-, beta- and gamma-carbonic anhydrases from bacteria: Can bacterial carbonic anhydrases shed new light on evolution of bacteria. J. Enzym. Inhib. Med. Chem. 2015, 30, 325–332.
- Supuran, C.T.; Capasso, C. The η-class carbonic anhydrases as drug targets for antimalarial agents. Expert Opin. Ther. Targets 2015, 19, 551–563.
- Del Prete, S.; Vullo, D.; De Luca, V.; Supuran, C.T.; Capasso, C. Biochemical characterization of the δ-carbonic anhydrase from the marine diatom Thalassiosiraweissflogii, TweCA. J. Enzym. Inhib. Med. Chem. 2014, 29, 906–911.
- Stefanucci, A.; Angeli, A.; Dimmito, M.P.; Luisi, G.; Del Prete, S.; Capasso, C.; Donald, W.A.; Mollica, A.; Supuran, C.T. Activation of β- and γ-carbonic anhydrases from pathogenic bacteria with tripeptides. J. Enzym. Inhib. Med. Chem. 2018, 33, 945–950.
- Angeli, A.; Del Prete, S.; Alasmary, F.A.S.; Alqahtani, L.S.; AlOthman, Z.; Donald, W.A.; Capasso, C.; Supuran, C.T. The first activation studies of the η-carbonic anhydrase from the malaria parasite Plasmodium falciparum with amines and amino acids. Bioorg. Chem. 2018, 80, 94–98.
- Angeli, A.; Buonanno, M.; Donald, W.A.; Monti, S.M.; Supuran, C.T. The zinc—But not cadmium—Containing ζ-carbonic from the diatom Thalassiosiraweissflogii is potently activated by amines and amino acids. Bioorg. Chem. 2018, 80, 261–265.
- Angeli, A.; Kuuslahti, M.; Parkkila, S.; Supuran, C.T. Activation studies with amines and amino acids of the α-carbonic anhydrase from the pathogenic protozoan Trypanosoma cruzi. Bioorg. Med. Chem. 2018, 26, 4187–4190.
- Angeli, A.; Del Prete, S.; Osman, S.M.; Alasmary, F.A.S.; AlOthman, Z.; Donald, W.A.; Capasso, C.; Supuran, C.T. Activation studies with amines and amino acids of the β-carbonic anhydrase encoded by the Rv3273 gene from the pathogenic bacterium Mycobacterium tuberculosis. J. Enzym. Inhib. Med. Chem. 2018, 33, 364–369.
- Angeli, A.; Del Prete, S.; Donald, W.A.; Capasso, C.; Supuran, C.T. The γ-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae is potently activated by amines and amino acids. Bioorg. Chem. 2018, 77, 1–5.
- Jensen, E.L.; Clement, R.; Kosta, A.; Maberly, S.C.; Gontero, B. A new widespread subclass of carbonic anhydrase in marine phytoplankton. ISME J. 2019, 13, 2094–2106.
- Nishimori, I.; Minakuchi, T.; Onishi, S.; Vullo, D.; Cecchi, A.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Cloning, characterization, and inhibition studies of the cytosolic isozyme III with sulfonamides. Bioorg. Med. Chem. 2007, 15, 7229–7236.
- Köhler, K.; Hillebrecht, A.; Schulze, W.J.; Innocenti, A.; Heine, A.; Supuran, C.T.; Klebe, G. Saccharin inhibits carbonicanhydrases: Possible explanation for its unpleasant metallic aftertaste. Angew. Chem. 2007, 46, 7697–7699.
- Supuran, C.T.; Scozzafava, A.; Ilies, M.A.; Briganti, F. Carbonic anhydrase inhibitors. Synthesis of sulfonamides incorporating 2,4,6-trisubstituted-pyridinium-ethylcarboxamido moieties possessing membrane-impermeability and in vivo selectivity for the membrane-bound (CA IV) versus the cytosolic (CA I and CA II) isozymes. J. Enzym. Inhib. 2000, 15, 381–401.
- Scozzafava, A.; Briganti, F.; Ilies, M.A.; Supuran, C.T. Carbonic anhydrase inhibitors. Synthesis of membrane-impermeant low molecular weight sulfonamides possessing in vivo selectivity for the membrane-bound versus the cytosolic isozymes. J. Med. Chem. 2000, 43, 292–300.
- De Simone, G.; Vitale, R.M.; Di Fiore, A.; Pedone, C.; Scozzafava, A.; Montero, J.L.; Winum, J.Y.; Supuran, C.T. Carbonic anhydrase inhibitors:hypoxia-activatable sulfonamides incorporatingdisulfide bonds that target the tumor-associatedisoform IX. J. Med. Chem. 2006, 49, 5544–5551.
- Maresca, A.; Temperini, C.; Vu, H.; Pham, N.B.; Poulsen, S.A.; Scozzafava, A.; Quinn, R.J.; Supuran, C.T. Non-zinc mediated inhibition of carbonic anhydrases: Coumarins are a new class of suicide inhibitors. J. Am. Chem. Soc. 2009, 131, 3057–3062.
- Maresca, A.; Temperini, C.; Pochet, L.; Masereel, B.; Scozzafava, A.; Supuran, C.T. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J. Med. Chem. 2010, 53, 335–344.
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug. Discov. 2008, 7, 168–181.
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032.
- Nocentini, A.; Supuran, C.T. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin. Drug Discov. 2019, 14, 1175–1197.
- Supuran, C.T. Coumarin carbonic anhydrase inhibitors from natural sources. J. Enzym. Inhib. Med. Chem. 2020, 35, 1462–1470.
- Wang, Z.C.; Qin, Y.J.; Wang, P.F.; Yang, Y.A.; Wen, Q.; Zhang, X.; Qiu, H.Y.; Duan, Y.T.; Wang, Y.T.; Sang, Y.L.; et al. Sulfonamides containing coumarin moieties selectively and potently inhibit carbonic anhydrases II and IX: Design, synthesis, inhibitory activity and 3D-QSAR analysis. Eur. J. Med. Chem. 2013, 66, 1–11.
- Kurt, B.Z.; Sonmez, F.; Bilen, C.; Ergun, A.; Gencer, N.; Arslan, O.; Kucukislamoglu, M. Synthesis, antioxidant and carbonic anhydrase I and II inhibitory activities of novel sulphonamide-substituted coumarylthiazole derivatives. J. Enzym. Inhib. Med. Chem. 2016, 31, 78–89.
- Bozdag, M.; Ferraroni, M.; Carta, F.; Vullo, D.; Lucarini, L.; Orlandini, E.; Rossello, A.; Nuti, E.; Scozzafava, A.; Masini, E.; et al. Structural insights on carbonic anhydrase inhibitory action, isoform selectivity, and potency of sulfonamides and coumarins incorporating arylsulfonylureido groups. J. Med. Chem. 2014, 57, 9152–9167.
- Chandak, N.; Ceruso, M.; Supuran, C.T.; Sharma, P.K. Novel sulfonamide bearing coumarin scaffolds as selective inhibitors of tumor associated carbonic anhydrase isoforms IX and XII. Bioorg. Med. Chem. 2016, 24, 2882–2886.

