Coumarin is an important six-membered aromatic heterocyclic pharmacophore, widely distributed in natural products and synthetic molecules. The versatile and unique features of coumarin nucleus, in combination with privileged sulfonamide moiety, have enhanced the broad spectrum of biological activities. The research and development of coumarin, sulfonamide-based pharmacology, and medicinal chemistry have become active topics, and attracted the attention of medicinal chemists, pharmacists, and synthetic chemists. Coumarin sulfonamide compounds and analogs as clinical drugs have been used to cure various diseases with high therapeutic potency, which have shown their enormous development value. The diversified and wide array of biological activities such as anticancer, antibacterial, anti-fungal, antioxidant and anti-viral, etc. were displayed by diversified coumarin sulfonamides.
- coumarin sulfonamide
- anticancer agents
1. Introduction
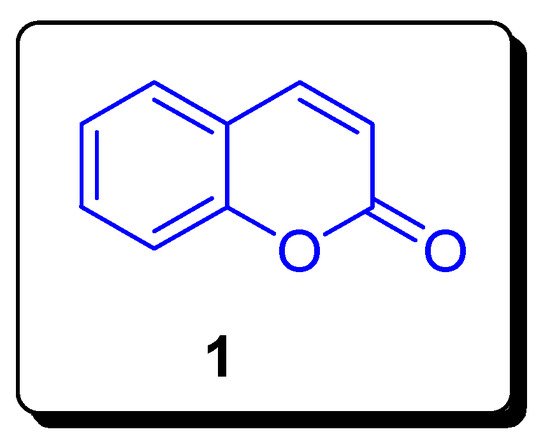
2. Coumarin Sulfonamides as Anti-Cancer Agents and Carbonic Anhydrase Inhibitors
2.1. Benzenesulfonamide-Based Coumarins as Carbonic Anhydrases II and IX Inhibitors
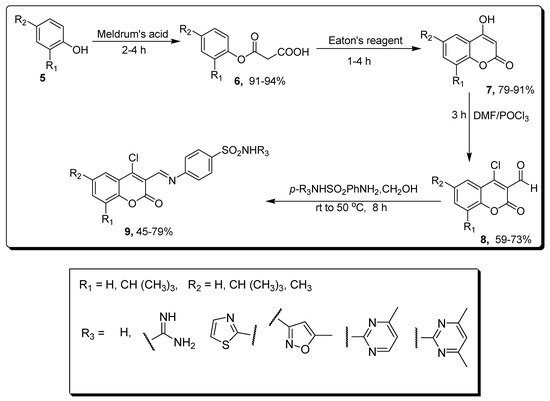
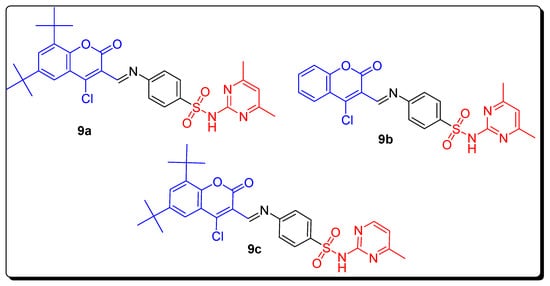
| Compound | MCF-7 µM | Compounds | hCAs II µM | hCAs IX µM |
|---|---|---|---|---|
| 9a | 0.0088 | 9b | - | 0.124 |
| Doxorubicin | 0.065 | 9c | 0.063 | - |
| Semaxanib | 0.0031 | AAZ | 0.016 | 0.028 |
| - | - | SA | 0.26 | 0.29 |
2.2. Thiazole-Sulfonamide Coumarin Hybrids as hCA I and hCA II Inhibitors
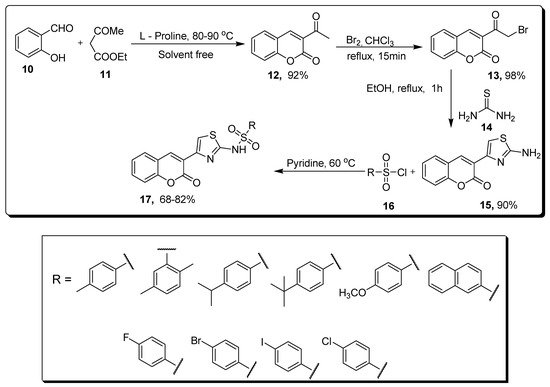
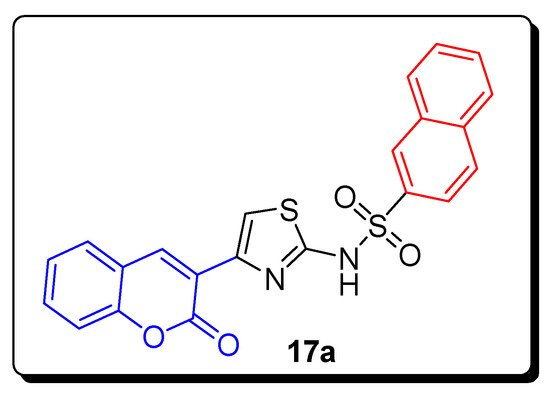
| Compound | hCA I IC | 50 | (µM) | hCA II IC | 50 | (µM) |
|---|---|---|---|---|---|---|
| 17a | 5.63 | 8.48 |
2.3. Sulfonyl Ureido Coumarins Hybrids as Carbonic Anhydrase Inhibitors
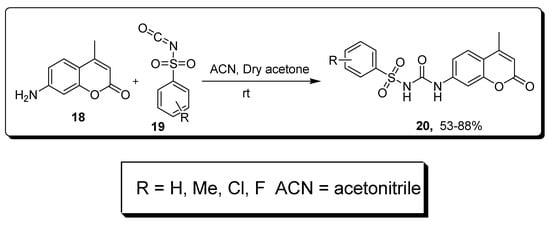
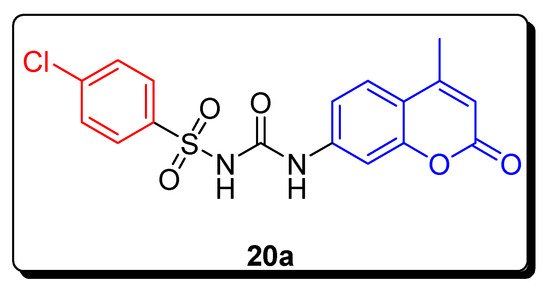
| Compound | hCA IX K | I | (nM) | hCA XII K | I | (nM) |
|---|---|---|---|---|---|---|
| 20a | 20.2 | 6.0 | ||||
| AAZ | 25.0 | 5.7 |
2.4. Benzene Sulfonamido-Coumarinyl Hydrazones Hybrids as CA Inhibitors
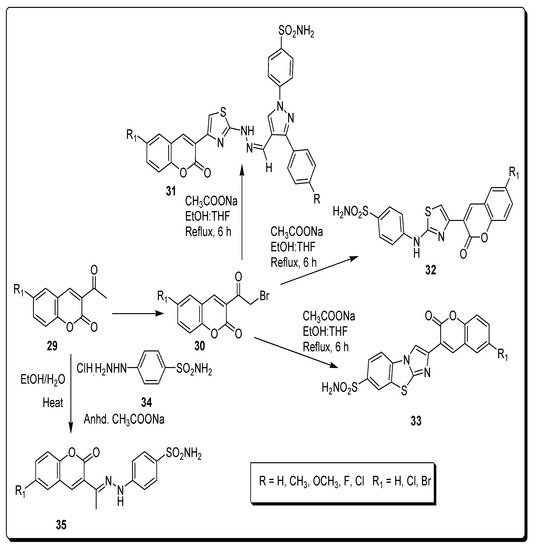
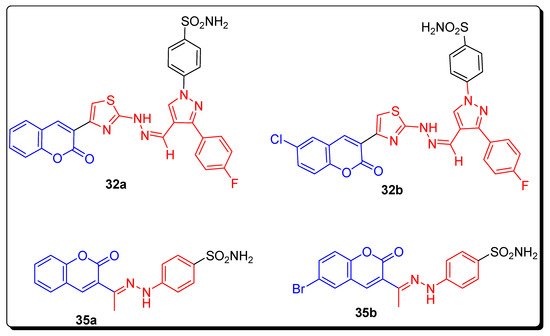
| Compound Number | hCA I K | I | (nM) | hCA II K | I | (nM) | hCA IX K | I | (nM) | hCA XII K | I | (nM) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 32a | 263.49 | 21.20 | 2.28 | 0.54 | ||||||||
| 32b | 349.63 | 17.46 | 2.54 | 0.54 | ||||||||
| 35a | 220.13 | 13.23 | 58.61 | 4.4 | ||||||||
| 35b | 21.95 | 1751.72 | 23.59 | 0.62 | ||||||||
| AZA | 250.0 | 12.1 | 25.0 | 5.7 |