 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Volker Schirrmacher | + 3101 word(s) | 3101 | 2022-03-09 06:56:37 | | | |
| 2 | Yvaine Wei | + 1 word(s) | 3102 | 2022-03-11 02:32:58 | | | | |
| 3 | Yvaine Wei | Meta information modification | 3102 | 2022-03-14 07:07:48 | | |
Video Upload Options
Oncolytic viruses represent interesting anti-cancer agents with high tumor selectivity and immune stimulatory potential. The anti-neoplastic activities of Newcastle Disease Virus (NDV) include (i) the endocytic targeting of the GTPase Rac1 in Ras-transformed human tumorigenic cells; (ii) the switch from cellular protein to viral protein synthesis and the induction of autophagy mediated by viral nucleoprotein NP; (iii) the virus replication mediated by viral RNA polymerase (large protein (L), associated with phosphoprotein (P)); (iv) the facilitation of NDV spread in tumors via the membrane budding of the virus progeny with the help of matrix protein (M) and fusion protein (F); and (v) the oncolysis via apoptosis, necroptosis, pyroptosis, or ferroptosis associated with immunogenic cell death. A special property of this oncolytic virus consists of its potential for breaking therapy resistance in human cancer cells.
1. Introduction
2. Basic Information on NDV
2.1. Genome
2.2. Cellular Infection and Viral Replication
- Cellular infection. In the case of membrane fusion, cellular infection starts with the virus binding to a host cell’s surface with α 2,6-linked sialic acid from glycoproteins or glycolipids via the cell-adhesion domain of HN [8]. This is followed by the activation of F. The concerted action of HN and F leads to conformational change, enabling fusion of the viral and the host cell membrane and thereby opening a pore to deliver the viral genome into the cytoplasm [9].
- Replication. When a sufficient amount of NP protein accumulates in the cytoplasm, a switch can occur from RNA transcription to replication. The polymerase complex then ignores the transcription stop signals at the 3′end of each gene and a full-length, positive-sense antigenome is synthesized. These antigenomic replicative intermediates are totally encapsidated by viral NP monomers, just like the full-length, negative-strand genomic RNA/ NP complex.
2.3. NDV Permissive and Non-Permissive Hosts
3. Intrinsic Anti-Neoplastic Activities
3.1. Targeting Rac1
3.2. Tumor-Selective Virus Replication
3.3. Tumor Selective Viral mRNA Translation
3.4. Tumor-Selective Shift to High Cytoplasmic and Cell Surface Expression of Viral Proteins
3.5. Tumor-Selective Switch from Positive Strand RNA Translation to Negative Strand Antigenome Synthesis
3.6. Tumor-Selective Switch to Autophagy
3.7. Tumor-Selective Oncolysis: Intrinsic Signaling Pathways
3.8. Oncolysis-Independent Effects
3.9. NDV Spread in Tumors
3.10. Breaking Cancer Therapy Resistancies
4. NDV-Modified Cancer Vaccine for Cancer Immunotherapy
4.1. Successful Application of NDV for Antimetastatic Active-Specific Immunotherapy (ASI)
4.2. Immunogenic Cancer Cell Death and Extrinsic Mechanisms of Oncolysis
4.3. Induction of Post-Oncolytic Immunity
4.4. Inhibition of Cell Proliferation by IFN-I
4.5. NDV Induced Upregulation of MHC I
4.6. Viral Immune Escape Mechanisms
4.7. NDV-Induced Interferon Response: Inhibition of Virus Replication
4.8. NDV-Induced Interferon Response: Induction of an Anti-Viral State
5. Immune Cell Activation
5.1. Activation of NK Cells, Monocytes, and Macrophages
5.2. Dendritic Cell Activation
5.3. Activation of T Cells
5.4. Oncolysis-Independent Immune Stimulatory Effects In Vivo
6. Schematic Diagram
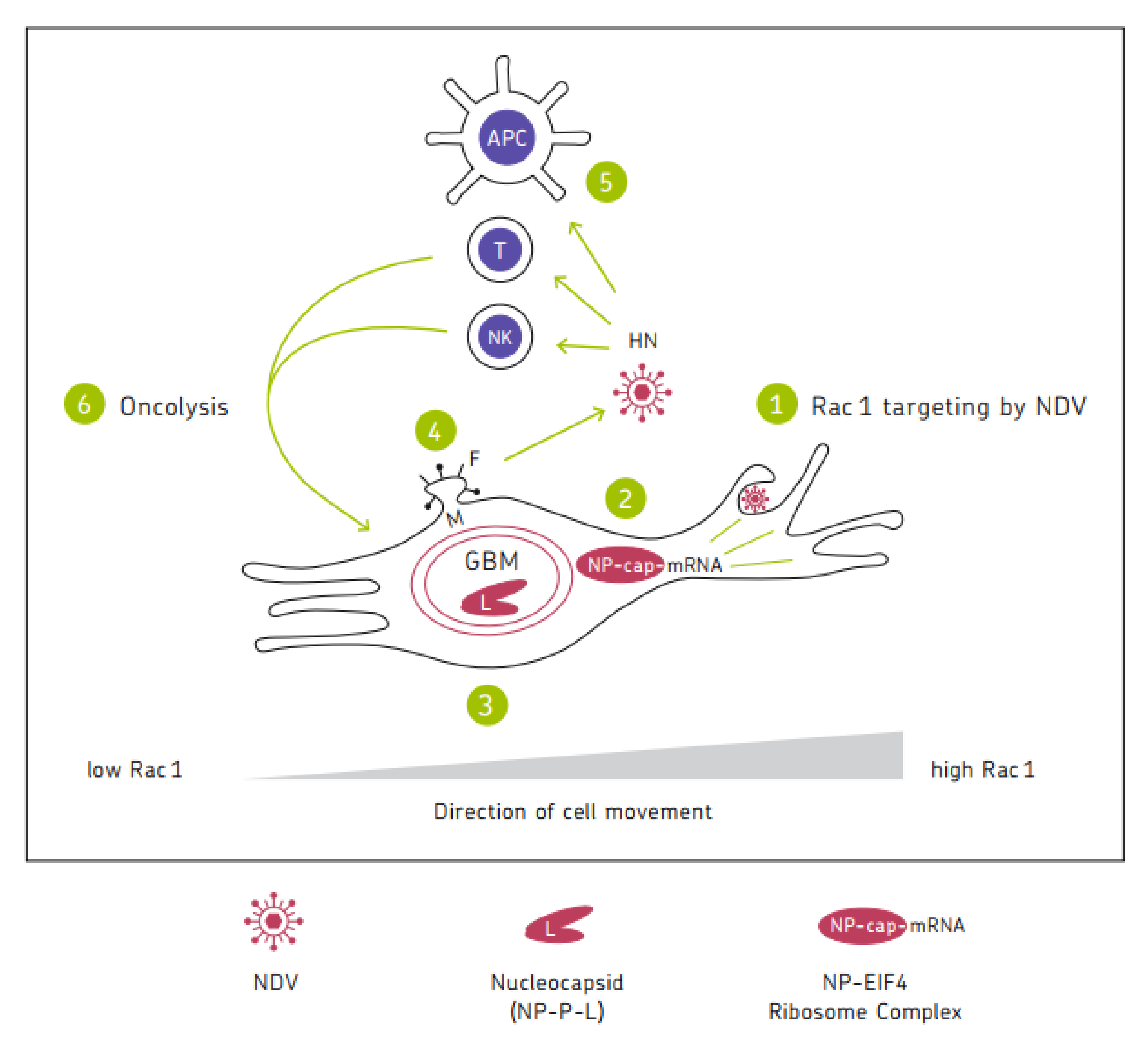
7. Conclusions
References
- Cao, G.-D.; He, X.-B.; Sun, Q.; Chen, S.; Wan, K.; Xu, X.; Feng, X.; Li, P.-P.; Chen, B.; Xiong, M. The Oncolytic Virus in Cancer Diagnosis and Treatment. Front. Oncol. 2020, 10, 1786.
- Cassel, W.A.; Garrett, R.E. Newcastle disease virus as an antineoplastic agent. Cancer 1965, 18, 863–868.
- Samal, S.K. Newcastle disease and related avian paramyxoviruses. In The Biology of Paramyxoviruses; Samal, S.K., Ed.; Caister Academic Press: Norfolk, UK, 2011; pp. 69–114.
- Song, X.; Shan, H.; Zhu, Y.; Hu, S.; Xue, L.; Chen, Y.; Ding, W.; Niu, T.; Gu, J.; Ouyang, S.; et al. Self-capping of nucleoprotein filaments protects the Newcastle disease virus genome. Elife 2019, 8, e45057.
- Nath, B.; Sharma, K.; Ahire, K.; Goyal, A.; Kumar, S. Structure analysis of the nucleoprotein of Newcastle disease virus: An insight towards its multimeric form in solution. Int. J. Biol. Macromol. 2020, 151, 402–411.
- Tan, L.; Zhang, Y.; Zhan, Y.; Yuan, Y.; Sun, Y.; Qiu, X.; Meng, C.; Song, C.; Liao, Y.; Ding, C. Newcastle disease virus employs macropinocytosis and Rab5a-dependent intracellular trafficking to infect DF-1 cells. Oncotarget 2016, 7, 86117–86133.
- El-Sayed, A.; Harashima, H. Endocytosis of Gene Delivery Vectors: From Clathrin-dependent to Lipid Raft-mediated Endocytosis. Mol. Ther. 2013, 21, 1118–1130.
- Yuan, P.; Paterson, R.G.; Leser, G.P.; Lamb, R.A.; Jardetzky, T.S. Structure of the Ulster Strain Newcastle Disease Virus Hemagglutinin-Neuraminidase Reveals Auto-Inhibitory Interactions Associated with Low Virulence. PLOS Pathog. 2012, 8, e1002855.
- Lamb, R.A.; Jardetzky, T.S. Structural basis of viral invasion: Lessons from paramyxovirus F. Curr. Opin. Struct. Biol. 2007, 17, 427–436.
- Puhlmann, J.; Puehler, F.; Mumberg, D.; Boukamp, P.; Beier, R. Rac1 is required for oncolytic NDV replication in human cancer cells and establishes a link between tumorigenesis and sensitivity to oncolytic virus. Oncogene 2010, 29, 2205–2215.
- Semenova, G.; Chernoff, J. Targeting PAK1. Biochem. Soc. Trans. 2017, 45, 79–88.
- Abdullah, J.M.; Mustafa, Z.; Ideris, A. Newcastle Disease Virus Interaction in Targeted Therapy against Proliferation and Invasion Pathways of Glioblastoma Multiforme. BioMed Res. Int. 2014, 2014, 386470.
- Mustafa, M.Z.; Shamsuddin, H.S.; Ideris, A.; Ibrahim, R.; Jaafar, R.; Ali, A.M.; Abdullah, J.M. Viability Reduction and Rac1 Gene Downregulation of Heterogeneous Ex-Vivo Glioma Acute Slice Infected by the Oncolytic Newcastle Disease Virus Strain V4UPM. BioMed Res. Int. 2013, 2013, 248507.
- Reichard, K.W.; Lorence, R.M.; Cascino, C.L.; Peeples, M.E.; Walter, R.J.; Fernando, M.B.; Reyes, H.M.; Greager, J.A. Newcastle disease virus selectively kills human tumor cells. J. Surg. Res. 1992, 52, 448–453.
- Phuangsab, A.; Lorence, R.M.; Reichard, K.W.; Peeples, M.E.; Walter, R.J. Newcastle disease virus therapy of human tumor xenografts: Antitumor effects of local or systemic administration. Cancer Lett. 2001, 172, 27–36.
- Kumar, R.; Khandelwal, N.; Thachamvally, R.; Tripathi, B.N.; Barua, S.; Kashyap, S.K.; Maherchandani, S.; Kumar, N. Role of MAPK/MNK1 signaling in virus replication. Virus Res. 2018, 253, 48–61.
- Zhan, Y.; Yu, S.; Yang, S.; Qiu, X.; Meng, C.; Tan, L.; Song, C.; Liao, Y.; Liu, W.; Sun, Y.; et al. Newcastle Disease virus infection activates PI3K/Akt/mTOR and p38 MAPK/Mnk1 pathways to benefit viral mRNA translation via interaction of the viral NP protein and host eIF4E. PLOS Pathog. 2020, 16, e1008610.
- Washburn, B.; Schirrmacher, V. Human tumor cell infection by Newcastle Disease Virus leads to upregulation of HLA and cell adhesion molecules and to induction of interferons, chemokines and finally apoptosis. Int. J. Oncol. 2002, 21, 85–93.
- Fiola, C.; Peeters, B.; Fournier, P.; Arnold, A.; Bucur, M.; Schirrmacher, V. Tumor selective replication of Newcastle disease virus: Association with defects of tumor cells in antiviral defence. Int. J. Cancer 2006, 119, 328–338.
- Cheng, J.-H.; Sun, Y.-J.; Zhang, F.-Q.; Zhang, X.-R.; Qiu, X.-S.; Yu, L.-P.; Wu, Y.-T.; Ding, C. Newcastle disease virus NP and P proteins induce autophagy via the endoplasmic reticulum stress-related unfolded protein response. Sci. Rep. 2016, 6, 24721.
- Urban-Wojciuk, Z.; Khan, M.M.; Oyler, B.L.; Fåhraeus, R.; Marek-Trzonkowska, N.; Nita-Lazar, A.; Hupp, T.R.; Goodlett, D.R. The Role of TLRs in Anti-cancer Immunity and Tumor Rejection. Front. Immunol. 2019, 10, 2388.
- Li, Y.; Jiang, W.; Niu, Q.; Sun, Y.; Meng, C.; Tan, L.; Song, C.; Qiu, X.; Liao, Y.; Ding, C. eIF2a-CHOP-BcL-2/JNK and IRE1a-XBP1/JNK signaling promote apoptosis and inflammation and support the proliferation of Newcastle disease virus. Cell Death Dis. 2019, 10, 891.
- Prabhu, S.A.; Moussa, O.; Miller, W.H., Jr.; Del Rincón, S.V. The MNK1/2-eIF4E Axis as a Potential Therapeutic Target in Melanoma. Int. J. Mol. Sci. 2020, 21, 4055.
- Shtykova, E.V.; Petoukhov, M.V.; Dadinova, L.A.; Fedorova, N.V.; Tashkin, V.Y.; Timofeeva, T.A.; Ksenofontov, A.L.; Loshkarev, N.A.; Baratova, L.A.; Jeffries, C.M.; et al. Solution Structure, Self-Assembly, and Membrane Interactions of the Matrix Protein from Newcastle Disease Virus at Neutral and Acidic pH. J. Virol. 2019, 93, e01450-18.
- Pantua, H.D.; McGinnes, L.W.; Peeples, M.E.; Morrison, T.G. Requirements for the Assembly and Release of Newcastle Disease Virus-Like Particles. J. Virol. 2006, 80, 11062–11073.
- Duan, Z.; Hu, Z.; Zhu, J.; Xu, H.; Chen, J.; Liu, H.; Hu, S.; Liu, X. Mutations in the FPIV motif of Newcastle disease virus matrix protein attenuate virus replication and reduce virus budding. Arch. Virol. 2014, 159, 1813–1819.
- Lamb, R.; Parks, G. In Paramyxoviridae: The viruses and their replication. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Roizman, B., Straus, S.E., Eds.; Lippencott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 1444–1496.
- Al-Ziaydi, A.G.; Al-Shammari, A.M.; Hamzah, M.I.; Kadhim, H.S.; Jabir, M.S. Newcastle disease virus suppress glycolysis pathway and induce breast cancer cells death. Virusdisease 2020, 31, 341–348.
- Schirrmacher, V.; Van Gool, S.; Stuecker, W. Breaking Therapy Resistance: An Update on Oncolytic Newcastle Disease Virus for Improvements of Cancer Therapy. Biomedicines 2019, 7, 66.
- Heicappell, R.; Schirrmacher, V.; von Hoegen, P.; Ahlert, T.; Appelhans, B. Prevention of metastatic spread by postoperative immunotherapy with virally modified autologous tumor cells. I. Parameters for optimal therapeutic effects. Int. J. Cancer 1986, 37, 569–577.
- Zangemeister-Wittke, U.; Kyewski, B.; Schirrmacher, V. Recruitment and activation of tumor-specific immune T cells in situ. CD8+ cells predominate the secondary response in sponge matrices and exert both delayed-type hypersensitivity-like and cytotoxic T lymphocyte activity. J. Immunol. 1989, 143, 379–385.
- Han, J.; Khatwani, N.; Searles, T.G.; Turk, M.J.; Angeles, C.V. Memory CD8+ T cell responses to cancer. Semin. Immunol. 2020, 49, 101435.
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized Oncolytic Virotherapy Overcomes Systemic Tumor Resistance to Immune Checkpoint Blockade Immunotherapy. Sci. Transl. Med. 2014, 6, 226ra32.
- Koks, C.A.; Garg, A.D.; Ehrhardt, M.; Riva, M.; Vandenberk, L.; Boon, L.; De Vleeschouwer, S.; Agostinis, P.; Graf, N.; Van Gool, S.W. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int. J. Cancer 2015, 136, E313–E325.
- Hong, M.; Zhang, Z.; Chen, Q.; Lu, Y.; Zhang, J.; Lin, C.; Zhang, F.; Zhang, W.; Li, X.; Zhang, W.; et al. IRF1 inhibits the proliferation and metastasis of colorectal cancer by suppressing the Ras-Rac1 pathway. Cancer Manag. Res. 2018, 11, 369–378.
- Baird, T.D.; Wek, R.C. Eukaryotic Initiation Factor 2 Phosphorylation and Translational Control in Metabolism. Adv. Nutr. 2012, 3, 307–321.
- Ten, R.M.; Blank, V.; Le Bail, O.; Kourilsky, P.; Israël, A. Two factors, IRF1 and KBF1/NF-kappa B, cooperate during induction of MHC class I gene expression by interferon alpha beta or Newcastle disease virus. C. R. Acad. Sci. III 1993, 316, 496–501.
- Schirrmacher, V. Signaling through RIG-I and type I interferon receptor: Immune activation by Newcastle disease virus in man versus immune evasion by Ebola virus (Review). Int. J. Mol. Med. 2015, 36, 3–10.
- Glosson, N.L.; Hudson, A.M. Human herpesvirus-6A and -6B encode viral immunoevasins that downregulate class I MHC molecules. Virology 2007, 365, 125–135.
- Koutsakos, M.; McWilliam, H.E.G.; Aktepe, T.E.; Fritzlar, S.; Illing, P.T.; Mifsud, N.A.; Purcell, A.W.; Rockman, S.; Reading, P.C.; Vivian, J.P.; et al. Downregulation of MHC Class I Expression by Influenza A and B Viruses. Front. Immunol. 2019, 10, 1158.
- Piguet, V. Receptor Modulation in Viral replication: HIV, HSV, HHV-8 and HPV: Same Goal, Different Techniques to Interfere with MHC-I Antigen Presentation. Curr. Top. Microbiol. Immunol. 2005, 285, 199–217.
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. Ser. B Boil. Sci. 1957, 147, 258–267.
- Lindemann, J. Viruses as immunological adjuvants in cancer. Biochim. Biophys. Acta 1974, 355, 49–75.
- Gresser, I.; Tovey, M.G.; Maury, C.; Bandu, M.T. Role of interferon in the pathogenesis of virus diseases in mice as demonstrated by the use of anti-interferon serum. II. Studies with herpes simplex, Moloney sarcoma, vesicular stomatitis, Newcastle disease, and influenza viruses. J. Exp. Med. 1976, 144, 1316–1323.
- Schirrmacher, V.; Fournier, P. Newcastle Disease Virus: A Promising Vector for Viral Therapy, Immune Therapy, and Gene Therapy of Cancer. In Methods in Molecular Biology, Gene Therapy of Cancer; Walther, W., Stein, U.S., Eds.; Humana Press: New York, NY, USA, 2009; Chapter 30; Volume 542, pp. 565–605.
- Wilden, H.; Fournier, P.; Zawatzky, R.; Schirrmacher, V. Expression of RIG-I, IRF3, IFN-beta and IRF7 determines resistance or susceptibility of cells to infection by Newcastle disease virus. Int. J. Oncol. 2009, 34, 971–982.
- Schirrmacher, V.; Fournier, P.; Wilden, H. Importance of retinoic acid-inducible gene I and of receptor for type I interferon for cellular resistance to infection by Newcastle disease virus. Int. J. Oncol. 2012, 40, 287–298.
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapeutic drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662.
- Hervas-Stubbs, S.; Perez-Gracia, J.L.; Rouzaut, A.; Sanmamed, M.F.; Le Bon, A.; Melero, I. Direct Effects of Type I Interferons on Cells of the Immune System. Clin. Cancer Res. 2011, 17, 2619–2627.
- Mahon, P.J.; Mirza, A.M.; Musich, T.A.; Iorio, R.M. Engineered Intermonomeric Disulfide Bonds in the Globular Domain of Newcastle Disease Virus Hemagglutinin-Neuraminidase Protein: Implications for the Mechanism of Fusion Promotion. J. Virol. 2008, 82, 10386–10396.
- Campbell, K.S.; Cohen, A.D.; Pazina, T. Mechanisms of NK cell activation and clinical activity of the therapeutic SLAM7 antibody, Elotuzumab in multiple myeloma. Front. Immunol. 2018, 9, 2551.
- Zaslavsky, E.; Hershberg, U.; Seto, J.; Pham, A.M.; Marquez, S.; Duke, J.L.; Wetmur, J.G.; Tenoever, B.R.; Sealfon, S.C.; Kleinstein, S.H. Antiviral Response Dictated by Choreographed Cascade of Transcription Factors. J. Immunol. 2010, 184, 2908–2917.
- Chapman, N.M.; Boothby, M.R.; Chi, H. Metabolic coordination of T cell quiescence and activation. Nat. Rev. Immunol. 2020, 20, 55–70.
- Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325–338.
- Azuma, M. Co-signal Molecules in T-Cell Activation: Historical overview and perspective. Adv. Exp. Med. Biol. 2019, 1189, 3–23.
- Yu, Y.; Smoligovets, A.A.; Groves, J.T. Modulation of T cell signaling by the actin cytoskeleton. J. Cell Sci. 2013, 126, 1049–1058.
- Oseledchyk, A.; Ricca, J.M.; Gigoux, M.; Ko, B.; Redelman-Sidi, G.; Walther, T.; Liu, C.; Iyer, G.; Merghoub, T.; Wolchok, J.D.; et al. Lysis-independent potentiation of immune checkpoint blockade by oncolytic virus. Oncotarget 2018, 9, 28702–28716.
- Apostolidis, L.; Schirrmacher, V.; Fournier, P. Host mediated anti-tumor effect of oncolytic Newcastle disease virus after locoregional application. Int. J. Oncol. 2007, 31, 1009–1019.
- Schirrmacher, V.; Griesbach, A.; Ahlert, T. Antitumor effects of Newcastle Disease Virus in vivo: Local versus systemic effects. Int. J. Oncol. 2001, 18, 945–952.
- Nguyen, L.K.; Kholodenko, B.N.; Von Kriegsheim, A. Rac1 and RhoA: Networks, loops and bistability. Small GTPases 2018, 9, 316–321.
- De, P.; Aske, J.C.; Dey, N. RAC1 Takes the Lead in Solid Tumors. Cells 2019, 8, 382.
- Liang, Y.; Song, D.Z.; Liang, S.; Zhang, Z.F.; Gao, L.X.; Fan, X.H. The hemagglutinin-neuraminidase protein of Newcastle disease virus upregulates expression of the TRAIL gene in murine natural killer cells through the activation of Syk and NF-kB. PLoS ONE 2017, 12, e0178746.

