Oncolytic viruses represent interesting anti-cancer agents with high tumor selectivity and immune stimulatory potential. The anti-neoplastic activities of Newcastle Disease Virus (NDV) include (i) the endocytic targeting of the GTPase Rac1 in Ras-transformed human tumorigenic cells; (ii) the switch from cellular protein to viral protein synthesis and the induction of autophagy mediated by viral nucleoprotein NP; (iii) the virus replication mediated by viral RNA polymerase (large protein (L), associated with phosphoprotein (P)); (iv) the facilitation of NDV spread in tumors via the membrane budding of the virus progeny with the help of matrix protein (M) and fusion protein (F); and (v) the oncolysis via apoptosis, necroptosis, pyroptosis, or ferroptosis associated with immunogenic cell death. A special property of this oncolytic virus consists of its potential for breaking therapy resistance in human cancer cells.
1. Introduction
An oncolytic virus (OV) is defined as an agent that can selectively replicate in and kill cancer cells without damaging healthy cells. An OV can be an attenuated naturally existing virus, or it can be a genetically engineered recombinant virus. Four OVs are presently approved for some clinical cancer treatment: Oncorine (H101), Rigvir, T-VEC [
1], and Delytact. The latter is a genetically engineered HSV-1 that was approved in Japan in 2021 for the treatment of patients with malignant glioma.
Cassel et al. first reported the anti-neoplastic and immune stimulatory properties of oncolytic avian Newcastle Disease Virus (NDV) in 1965 [
2]. Because of its oncolytic and pluripotent immune stimulatory properties, NDV has become an agent of worldwide research interest. New findings provide a deep insight into mechanisms of tumor-selective oncolysis and immune stimulation.
2. Basic Information on NDV
2.1. Genome
The genome of NDV is a non-segmented, negative-sense, single-stranded (ss) RNA of slightly less than 15200 nucleotides (nt). All the genome sizes of NDV are consistent with the “rule of six”, which is typical for Paramyxoviridae. This rule is thought to arise from the need to fully encapsidate the entire length of the genome with a chain of NP monomers that span exactly 6 nt. The nucleocapsides of the most prevalent genome-length NDV strains contain about 2530 NP monomers [
4]. The genome is encapsidated by NP self-assembly into tightly packed helical 28 nm-diameter nucleocapsids with a clam-shaped structure which functions for genome transcription and replication [
5,
6].
2.2. Cellular Infection and Viral Replication
Infection of cells by NDV can occur via membrane fusion [
4,
5] or via endocytosis. In the case of endocytosis, NDV can use various pathways: clathrin-mediated endocytosis in non-lipid raft membrane domains [
8], phagocytosis and macropinocytosis in mixed membrane domains [
8], and RhoA-dependent endocytosis in lipid raft membrane domains [
9].
It can schematically be divided into two steps:
- Cellular infection. In the case of membrane fusion, cellular infection starts with the virus binding to a host cell’s surface with α 2,6-linked sialic acid from glycoproteins or glycolipids via the cell-adhesion domain of HN [10]. This is followed by the activation of F. The concerted action of HN and F leads to conformational change, enabling fusion of the viral and the host cell membrane and thereby opening a pore to deliver the viral genome into the cytoplasm [11].
- Replication. When a sufficient amount of NP protein accumulates in the cytoplasm, a switch can occur from RNA transcription to replication. The polymerase complex then ignores the transcription stop signals at the 3′end of each gene and a full-length, positive-sense antigenome is synthesized. These antigenomic replicative intermediates are totally encapsidated by viral NP monomers, just like the full-length, negative-strand genomic RNA/ NP complex.
2.3. NDV Permissive and Non-Permissive Hosts
The permissive hosts of NDV are birds. NDV is widespread in many countries worldwide and can infect over 250 bird species. The non-permissive hosts of NDV are all vertebrates except birds.
Class II NDV strains are diverse, with at least 20 genotypes. They include most oncolytic strains of medical interest, e.g., lentogenic Ulster, B1 and La Sota, mesogenic Mukteswar and velogenic Italien, and Hert33.
3. Intrinsic Anti-Neoplastic Activities
Anticancer chemotherapeutic drugs with their relatively low level of tumor selectivity exert unwanted non-target side effects. New agents with higher tumor selectivity are therefore urgently needed. As oncolytic NDV has high tumor selectivity in humans, it is important to unravel the molecular mechanisms of this agent.
3.1. Targeting Rac1
The small Rho GTPase Rac1 is targeted by NDV in human Ras-transformed tumorigenic cell lines. In these cells, Ras is activated via membrane growth factor receptors (e.g., EGFR, PDGFR) [
19]. Rac1 activates Pak1 which plays a central role in many oncogenic signaling pathways in human cells [
20]. NDV interacts with Rac1 in transformed cells upon viral entry, during syncytium induction, and upon actin reorganization. This OV is therefore recommended for targeted therapy against the proliferation and invasion pathways of glioblastoma multiforme (GBM) [
21]. Rac1 gene downregulation led to the inhibition of NDV replication [
22].
3.2. Tumor-Selective Virus Replication
Oncolytic NDV can replicate up to 10,000-fold in human cancer cells [
23]. It selectively kills tumor cells in vitro [
23] and exerts antitumor effects in vivo in human tumor xenografts [
24]. In contrast, oncolytic NDV does not replicate in and kill human non-transformed cells.
The reasons for this difference in susceptibility to NDV virus replication between tumorigenic and non-tumorigenic cells are multifold and will be discussed before going into the molecular details.
3.3. Tumor Selective Viral mRNA Translation
In Ras-transformed tumor cells, the p38 mitogen-activated protein kinase (MAPK) pathway (Ras/Raf/MEK/ERK) is activated by the binding of receptor ligands to the cognate receptor tyrosine kinase. With the help of adaptor proteins, the activated receptors convert Ras-GDP to Ras-GTP. The information is further transferred by phosphorylation (activation) of a series of enzymes (Raf, MEK1/2, ERK1/2), which leads to the activation of MAPK-interacting kinase 1 (Mnk1). Activated Mnk1 leads to dissociation of the 4EBP1-eIF4E complex, which results in the release, phosphorylation, and activation of the mRNA cap-binding eucaryotic translation initiation factor eIF4E [
34]. The Mnk1/2-eIF4E axis is often dysregulated in cancer and recommended as a potential therapeutic target in melanoma [
35].
3.4. Tumor-Selective Shift to High Cytoplasmic and Cell Surface Expression of Viral Proteins
The NDV infection of human tumor cells, irradiated by 200 Gray (Gy), was demonstrated by FACS flow cytometry to lead to a shift towards the high-density cell surface expression of viral HN and F molecules [
37]. Such shifts were not observed with nontumorigenic human cells.
Pronounced differences between human tumor cell lines and nontumorigenic cells were also observed upon infection by a recombinant lentogenic strain of NDV (Ulster) expressing an incorporated marker transgene (enhanced green-fluorescent protein, EGFP) (NDFL-EGFP). A long-lasting strong cytoplasmic EGFP fluorescence signal was observed in the whole cell population of tumor cells. In nontumorigenic cells, the fluorescence signals were only weak or missing completely [
32].
3.5. Tumor-Selective Switch from Positive Strand RNA Translation to Negative Strand Antigenome Synthesis
A comparative analysis of human tumorigenic and nontumorigenic cells revealed that within 1 h of infection by NDV, the amount of positive-strand RNA increases in both cell types. In tumor lines, an increase in negative-strand RNA was observed after about 12 h. In contrast, in nontumorigenic cells the expression of negative-strand RNA remained weak and transitory [
32].
3.6. Tumor-Selective Switch to Autophagy
More detailed information concerning NDV-mediated oncolysis became available within the last 15 years. This includes the unfolded protein ER stress response (UPR), UPR signaling, autophagy, and apoptosis [
44].
ER plays an important role in regulating protein synthesis/processing, lipid synthesis, and calcium homeostasis. During virus infection, many viral proteins are synthesized by ER-associated ribosomes and transported into the ER lumen for proper folding or posttranslational modification.
3.7. Tumor-Selective Oncolysis: Intrinsic Signaling Pathways
If ER homeostasis cannot be restored, UPR drives the damaged or infected cells to apoptosis [
43]. eIF2α-CHOP-BcL-2/JNK and IRE1a-XBP1/JNK signaling was reported to promote apoptosis and inflammation and to support the proliferation of NDV [
45]. BcL-2 is a mitochondrial anti-apoptotic protein. Knock down and overexpression studies showed that C/EBP-homologous protein (CHOP), IRE1α, X-box binding protein 1 (XBP1), and jun kinase (JNK) support efficient virus proliferation. The cellular translation shut-off caused by PERK/PKR-eIF2α signaling [
32,
45] and the hacking of the translational machinery by NDV infection via NP-eIF4E interaction [
36] allow NDV-infected tumor cells to translate the viral proteins preferentially.
3.8. Oncolysis-Independent Effects
Oncolysis-independent anti-tumor effects of NDV also exist. They have to do with improved adhesiveness between virus-infected tumor cells and cells from the immune system, such as NK and T cells. These immune stimulatory and co-stimulatory aspects will be reported below.
3.9. NDV Spread in Tumors
Following virus replication, NDV assembles and buds from the plasma membranes of the infected cells. In this process, M proteins play an important role. They have the potential to self-assemble into hollow helical oligomers [
13] and to interact with viral NP and HN proteins and also with cellular multivesicular body (MVB) proteins [
57,
58].
The release of progeny virions from the surface of infected cells is facilitated by neuraminidase activity located at the sialidase ß-propeller domain [
10] of the HN protein. This cleaves sialic acid from sugar side chains, thereby releasing the progeny virions [
59].
3.10. Breaking Cancer Therapy Resistancies
So far, it was reported that the anti-neoplastic effects of NDV in humans include (i) targeting the oncogenic protein Rac1, (ii) replicating selectively in tumor cells via autophagy, (iii) selectively destroying tumor cells (viral oncolysis), and (iv) promoting virus spread via syncytia and exosomes. NDV can also suppress the glycolysis pathway, which is an important energy source for cell growth and proliferation [
71].
Beyond these anti-neoplastic effects, oncolytic NDV has the intrinsic potential to break the resistance of cancer cells to a variety of therapies [
27].
4. NDV-Modified Cancer Vaccine for Cancer Immunotherapy
4.1. Successful Application of NDV for Antimetastatic Active-Specific Immunotherapy (ASI)
The prevention of metastatic spread was reported by postoperative active-specific immunotherapy (ASI) with NDV virally modified but not with unmodified irradiated autologous tumor cells in the ESb mouse tumor model [
87]. This demonstrated a successful application of NDV for antimetastatic cancer immunotherapy. Associated studies in this model provided the earliest evidence for the generation of protective immune T-cell-mediated memory responses to cancer [
88]. A recent review describes how cancer-reactive memory CD8+ T cell subsets orchestrate durable immunity to cancer [
89].
4.2. Immunogenic Cancer Cell Death and Extrinsic Mechanisms of Oncolysis
The in situ activation of protective T cells can occur not only by post-operative immunization with a virus-modified cancer vaccine [
87] but also by the direct inoculation of oncolytic NDV into primary tumors. Zamarin and colleagues demonstrated in 2014 that localized oncolytic virotherapy with NDV overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy [
84]. This was interpreted as being based on the induction of immunogenic cell death (ICD) and of systemic protective immunity leading to an abscopal effect at the site of a second untreated tumor.
4.3. Induction of Post-Oncolytic Immunity
The induction of post-oncolytic immunity by the intratumoral application of oncolytic NDV was analyzed in the above-mentioned orthotopic mouse glioma model in immunocompetent mice [
94]. The therapeutic effects observed relied on the induction of ICD and on the induction of adaptive T-cell-mediated anti-tumor immunity. The NDV-treated tumors became infiltrated by T cells producing interferon gamma (IFN–γ). In immunodeficient T cell receptor V(D)J recombinase 2 knockout (RAG2
−/−) mice or mice depleted of CD8+ T cells, no therapeutic effects were seen [
94].
4.4. Inhibition of Cell Proliferation by IFN-I
Human type I interferons are a group of interferon proteins that regulate cell growth and help to regulate the activity of the immune system.
IFN-I and retinoic acid (RA) are known to inhibit the proliferation of many normal and transformed cells. Both have in vivo antitumor activity. Induction of the transcription factor (TF) IFN regulatory factor-1 (IRF-1) was reported to inhibit colorectal cancer proliferation and metastasis by suppressing the Ras-Rac1 pathway [
101]. IRF-1 functions as a tumor suppressor. IRFs bind to the IFN promoters of specific target genes and induce the expression of IFN when tissues are infected.
4.5. NDV Induced Upregulation of MHC I
The NDV infection of human cancer cells leads to the upregulation of major histocompatibility complex (MHC) class I molecules (HLA) [
38]. This can be explained by the cooperative interactions of two induced TFs, IRF-1, which binds to the interferon response sequence of the MHC I gene promoter, and NFκB, which binds to the respective enhancer region [
102].
4.6. Viral Immune Escape Mechanisms
Viruses in their permissive hosts develop immune escape mechanisms that interfere with the IFN-I response and/or with proper MHC I-mediated antigen presentation. NDV in birds and Ebola virus in primates target and inhibit the IFN-I response [
103]. Many human pathogenic viruses, such as herpesvirus, influenza virus, and HIV, target and downregulate MHC I-mediated antigen presentation to escape adaptive T-cell-mediated immune responses [
104,
105,
106].
4.7. NDV-Induced Interferon Response: Inhibition of Virus Replication
IFN-I was discovered in 1957 by Isaacs and Lindenmann as a factor capable of interference with viruses [
107]. Later, in 1974, Lindenmann suggested that viruses could be used as immunological adjuvants in cancer, based on his studies on viral oncolysis and post-oncolytic immunity [
108]. In 1976, Gresser and colleagues demonstrated the importance of the early production of IFN-I in the response of the mouse to several viruses, including NDV [
109].
4.8. NDV-Induced Interferon Response: Induction of an Anti-Viral State
In mammalian cells, NDV induces a strong type I interferon response [
7,
27,
28,
29]. This involves an early and a late phase and leads to the inhibition of virus replication.
In short, the early phase is initiated by cytoplasmic viral RNA-activating PKR and the RIG-I/mitochondrial antiviral signaling protein (MAVS) pathway. In the late phase, the released interferons α and ß initiate an amplification loop by activating IFNAR [
29,
30].
5. Immune Cell Activation
When interferon was discovered, its function of virus interference was the first effect seen. Meanwhile, it is well established that type I IFNs exert direct effects not only on viruses but also on the cells of the immune system [
116].
5.1. Activation of NK Cells, Monocytes, and Macrophages
The HN protein of NDV directly binds to the activating receptors NKp46 and NKp44 of murine NK cells. These two receptors facilitate signal transduction and NK cell activation via the immunoreceptor tyrosine-based activation motif (ITAM)-linked signaling chains CD3ζ (NKp46) and DAP12 (NKp44) [
101]. The HN protein on infected cell membranes exists as a tetramer composed of a pair of dimers [
117]. These multimer ligands lead to NK cell activation via cross-linking their activation receptors [
118].
5.2. Dendritic Cell Activation
Of importance for the induction of adaptive T-cell-mediated immunity is the correct means of DC activation.
Human myeloid dendritic cells (mDCs) were infected by NDV to study a cellular response to virus infection that is not inhibited by any viral escape mechanism. A new approach of systems biology integrated genome-wide expression kinetics and time-dependent promoter analysis. Within 18 h, the cells established an anti-viral state. This was explained by a convergent regulatory network of transcription factors [TFs]. A network of 24 TFs was predicted to regulate 779 of the 1351 upregulated genes. The effect of NDV on mDCs was highly reproducible. The timing of this step-wise transcriptional signal propagation appeared as highly conserved [
123].
5.3. Activation of T Cells
Naïve T cells are maintained in a quiescent state that promotes their survival and persistence [
134]. Antigen recognition by naïve CD4+ and CD8+ T cells triggers mTOR activation which programs their differentiation into functionally distinct lineages [
135]. Co-stimulatory and co-inhibitory receptors and ligands fine-tune various immune responses [
136]. T cell signaling is also modulated by metabolic coordination [
134] and by the actin cytoskeleton [
137].
5.4. Oncolysis-Independent Immune Stimulatory Effects In Vivo
Virus potentiation of tumor vaccine T cell stimulatory capacity was reported to require cell surface binding but not infection [
71]. This can explain why therapeutic effects in vivo can be obtained with NDV even against in vitro oncolysis-resistant tumor lines [
85,
149].
In a human melanoma nude mouse xenotransplant model, strong anti-tumor bystander effects were observed by a 20% co-admixture of melanoma cells pre-infected in vitro by lentogenic NDV (Ulster), suggesting oncolysis-independent innate immunity activation [
150].
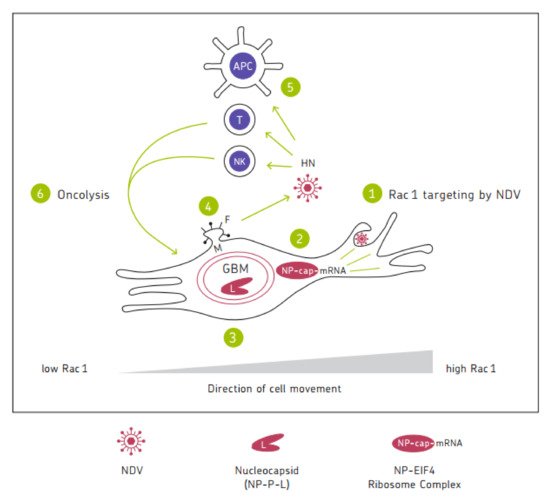
6. Schematic Diagram
Figure 1 shows a chart illustrating the mechanisms of the anti-tumor activity of oncolytic NDV. It represents a cycle of six steps in which the six viral proteins are involved. Mesogenic or velogenic NDV strains with their multicyclic replication capacity have the potential to drive several rounds of the cycle, thereby increasing the intensity and duration of the anti-cancer immune response.
Figure 1. Schematic illustration of mechanisms of anti-tumor activity of NDV. This is exemplified with a migratory and invasive glioblastoma (GBM) cell. The direction of cell movement is accompanied by an increase in Rac1 expression from the trailing edge (
left) to the leading edge at the lamellipodia (
right) [
151]. 1. Macropinocytosis/endocytosis of NDV-targeting Rac1, which plays a cardinal role in oncogenic alterations and in the development of drug resistance [
152]. The direction of cell movement is accompanied by an increase in Rac1 expression from the trailing edge (
left) to the leading edge at the lamellipodia (
right) [
151]. 2. Targeting the cap-dependent translational machinery; viral mRNA to protein translation in the cytosol and later in the double-membraned autophagosome (see GBM). This is achieved through the MNK1/2-eIF4E axis [
35,
36]. 3. Tumor-selective virus replication in autophagosomes. The clam shaped symbol stands for the helical nucleocapsid structure composed of L, P, and NP proteins [
5,
6]. 4. Virus progeny encapsulation, budding, and virus release mediated via M, HN, and F. 5. Antiviral response of healthy normal cells, including immune cells (APC, T, NK) initiated through recognition of HN by NKp46 and other PAMPs by TLRs and RLRs leading to IFN-I secretion and to DC1 and Th1 polarized adaptive immunity responses. 6. NDV-induced tumor cell death responses (oncolysis). These involve extrinsic (immune-mediated) and intrinsic cell death signaling pathways [
98]. ICD-derived components feed into APCs which present TAAs to T cells. Several rounds of such cycles (1–6) drive oncolytic effects and lead to immunological memory and systemic antitumor immunity.
7. Conclusions
As biological agents, OVs can replicate in tumor tissue and thereby augment their therapeutic effect. Tumor selectivity of this phenomenon ensures that healthy tissue is spared and that side effects are low.
This review reports milestones in the discovery of cellular and molecular mechanisms involved in tumor-selective virus replication and oncolysis and the resistance of normal healthy cells and of immune stimulatory effects.
It can be concluded that viral structural proteins from avian NDV successfully interact with proteins from non-permissive mammalian cells. This is not self-evident, considering the fact that the viral non-structural protein V antagonizes IFN α,ß signaling in cells from birds but not from humans. Avian NP interacts with mammalian eIF4E protein in viral mRNA translation and thus competes for binding with the corresponding cellular protein in the translation initiation complex. NP and P proteins induce autophagy in human cancer cells. M proteins interact during virus self-assembly with cellular MVB proteins. HN proteins interact with human NK-cell-activating receptors, co-stimulate T cells, and induce IFN-I responses. In addition, viral mRNAs interact with ribosomal proteins during protein translation, and ppp-RNA Leader interacts with RIG-I helicase. This latter interaction activates a cellular anti-viral defense signaling pathway. In contrast, NDV interacting with Rac1 GTPase in the lamellipodia of invasive cancer cells inactivates the cellular oncogenic signaling pathways [
19].
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10030562