Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kwang-Hyeok Kim | + 570 word(s) | 570 | 2022-02-24 03:56:41 | | | |
| 2 | Kwang-Hyeok Kim | + 2070 word(s) | 2640 | 2022-03-03 14:18:59 | | | | |
| 3 | Rita Xu | -132 word(s) | 2508 | 2022-03-04 04:18:56 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kim, K. Antibody-Drug Conjugate Targeting c-Kit. Encyclopedia. Available online: https://encyclopedia.pub/entry/20166 (accessed on 26 July 2026).
Kim K. Antibody-Drug Conjugate Targeting c-Kit. Encyclopedia. Available at: https://encyclopedia.pub/entry/20166. Accessed July 26, 2026.
Kim, Kwang-Hyeok. "Antibody-Drug Conjugate Targeting c-Kit" Encyclopedia, https://encyclopedia.pub/entry/20166 (accessed July 26, 2026).
Kim, K. (2022, March 03). Antibody-Drug Conjugate Targeting c-Kit. In Encyclopedia. https://encyclopedia.pub/entry/20166
Kim, Kwang-Hyeok. "Antibody-Drug Conjugate Targeting c-Kit." Encyclopedia. Web. 03 March, 2022.
Copy Citation
Lung cancer is the leading cause of cancer-related deaths. Small cell lung cancer (SCLC) accounts for 15–25% of all lung cancers. It exhibits a rapid doubling time and a high degree of invasiveness. Additionally, overexpression of c-Kit occurs in 70% of SCLC patients.
c-Kit
small cell lung cancer
monoclonal antibody
antibody-drug conjugate
1. Introduction
Lung cancer is the leading cause of cancer-related deaths in the Western world and is classified into two groups: small cell lung cancer (SCLC) and non-SCLC (NSCLC) [1]. SCLC, a neuroendocrine tumor, is distinguished from NSCLC by its rapid tumor growth, high degree of invasiveness and early development of widespread metastases [2]. SCLC is distinctly different from extrapulmonary small cell carcinoma with respect to disease progression, prognosis, and etiology [3]. Without proper treatment, the life expectancy of SCLC patients is less than four months. Although the five-year relative survival rate has improved by 7% over the last few decades, it remains extremely poor [4].
A variety of molecular markers have been implicated in the pathogenesis and prognosis of SCLC [5][6]. Paracrine or autocrine signal transduction pathways are widely used to explain dysregulated SCLC growth [6]. In addition, tumor protein p53, retinoblastoma protein, NOTCH, MYC, and phosphatidylinositol 3-kinase (PI3K) are aberrantly mutated in SCLC; however, well-established etiological factors, such as EGFR mutations that occur in NSCLC, have not been identified [7][8][9][10]. SCLC has a very aggressive course and is characterized by genomic instability, increased vascularity, and a high metastatic potential [11]. Consequently, most SCLC patients already present with metastatic disease outside of the chest, at the time of diagnosis, which results in premature death [12]. In addition, most SCLC patients are current or former heavy smokers, which is associated with a high tumor mutational burden, with C:G > A:T transversions being the most common type of base substitutions [13][14]
The c-Kit proto-oncogene encodes a transmembrane tyrosine kinase growth factor receptor that belongs to the platelet-derived growth factor receptor (PDGFR) family [15][16]. Its ligand stem cell factor (SCF) is a hematopoietic growth factor that promotes the proliferation of multiple hematopoietic stem cells [17][18]. In addition, c-Kit activity is dysregulated in various cancers [19][20]. Previous studies reported that the expression of c-Kit containing oncogenic mutations is either dysregulated and/or up-regulated in various cancers, which results in SCF-independent c-Kit activation and an aggressive form of cancer [20]. Interestingly, a variety of evidence indicates that SCLC cell lines and tumors express both the c-Kit receptor and SCF mRNA, suggesting that these gene products constitute an autocrine loop that mediates tumor cell survival and growth [21][22]. Although SCLC is significantly correlated with smoking, it does not contain oncogenic c-Kit mutations. Immunohistochemical staining showed that overexpression of c-Kit occurs in 70% of SCLC patients [23][24]. Imatinib, which was developed to target BCR-ABL, platelet-derived growth factor receptor (PDGFR), and c-Kit, is currently used to treat chronic myeloid leukemia, acute lymphoid leukemia, gastrointestinal stromal tumor (GIST), and hypereosinophilic syndrome [25][26][27][28]. A variety of in vitro and in vivo studies demonstrated that imatinib exhibits therapeutic efficacy against SCLC [29][30]. However, in phase 2 clinical trials, imatinib failed to exhibit significant therapeutic efficacy as shown by a lack of objective responses [31][32][33]. Thus, an alternative approach to target c-Kit in SCLC is needed.
2. 4C9 Antibody Specifically Binds to c-Kit
First, researchers examined whether the 4C9 antibody specifically binds to c-Kit on cell surface. FACS analysis revealed that 4C9 binds to various SCLC cell lines, including NCI-H526, NCI-H1048, and NCI-H889, in a dose-dependent manner (Figure 1A). Interestingly, 4C9 antibody binding was saturated at 50 ng/mL in c-Kit-positive SCLC cell lines. In addition, the expression of c-Kit was higher in NCI-H889 cells compared with that in NCI-H526 and NCI-H1048 cells, which is consistent with a previous report [34]. However, 4C9 did not show cross-reactivity with NCI-H446 and NCI-H2170 cells, which are c-Kit-negative SCLC cell lines. Researchers further examined the specific binding of 4C9 to c-Kit using siRNA knockdown experiment. Western blot analysis showed that c-Kit siRNA efficiently decreased protein expression in NCI-H1048 cells (Figure 1B). FACS analysis demonstrated that c-Kit expression on the cell surface was also down-regulated by c-Kit siRNA. Taken together, 4C9 binds specifically to the extracellular domain of c-Kit on the cell surface.
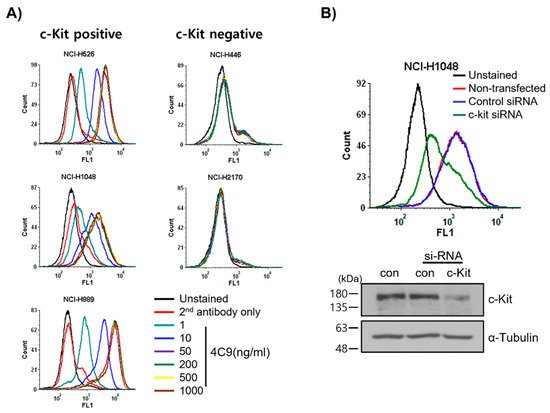
Figure 1. Determination of specific binding of the 4C9 antibody to the surface of SCLC cells. (A) SCLC cell lines were incubated with 4C9 antibody in a dose-dependent manner and analyzed by FACS. (B) NCI-H1048 cells were transfected with control or c-Kit si-RNA and incubated for 72 h. Binding of 4C9 (1 μg/mL) to NCI-H1048 cells was determined by FACS. The si-RNA-mediated down-regulation of c-Kit protein expression was evaluated by Western blot analysis. Alpha-tubulin was used as a loading control.
Next, researchers investigated whether the 4C9 antibody could inhibit the binding of SCF, a ligand for c-Kit. Competitive enzyme-linked immunosorbent assay (ELISA) results showed that the binding of 4C9 antibody to c-Kit was not affected by SCF, even at high concentrations (Figure 2A), suggesting that 4C9 antibody binds to c-Kit independent of SCF. In addition, researchers assessed whether 4C9 could inhibit SCF-mediated phosphorylation of c-Kit. Using GIST-T1 cells, pretreatment with 4C9 antibody resulted in decreased c-Kit phosphorylation in a dose-dependent manner; however, the phosphorylation levels of the ERK and Akt, downstream molecules of c-Kit, were not changed by treatment with 4C9 antibody (Figure 2B). Interestingly, total c-Kit levels decreased by 4C9 antibody treatment (Figure 2B). Hence, a decrease in phosphorylated c-Kit levels may result from decreased expression of c-Kit. A stability assessment of c-Kit indicated that the 4C9 antibody dramatically decreased total c-Kit levels in a time-dependent manner in both GIST cell lines and in some SCLC cell lines, which may be associated with ubiquitination-dependent degradation. Nevertheless, this needs further elucidation. In contrast to GIST-T1, 4C9 did not reduce SCF-mediated c-Kit phosphorylation or c-Kit stability in the NCI-H526 and NCI-H1048 cell lines (Figure 2C). Furthermore, the 4C9 antibody did not inhibit phosphorylation of ERK and Akt induced by SCF, which suggests that the 4C9 antibody does not function as an antagonist of SCF/c-Kit signaling.
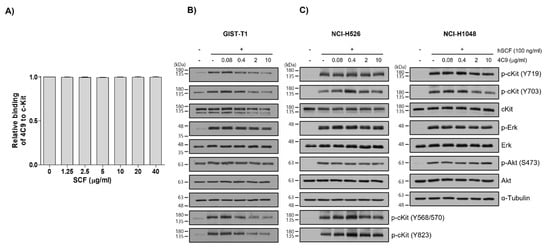
Figure 2. Characterization of the 4C9 antibody. (A) Human c-Kit (20 ng/well) was coated onto 96-well plates and the binding of the 4C9 antibody was investigated in the presence of human SCF at the indicated concentrations. The results represent the mean ± SD of three independent experiments. (B,C) GIST-T1, NCI-H526, or NCI-H1048 cells were treated with 4C9 at the indicated concentrations in the presence or absence of SCF (100 ng/mL). The phosphorylation of c-Kit, Akt, and ERK was assessed by Western blot analysis. In NCI-H1048 cells, phosphorylation of Y568/570 and Y823 by SCF treatment was not detected. Alpha-tubulin was used as a loading control. The results represent the mean ± SD of three independent experiments.
3. Generation and Characterization of ADC (4C9-DM1)
Even though c-Kit is overexpressed in SCLC cell lines, naked antibodies cannot be applied to treat SCLC because of the limited contribution of SCF/c-Kit signaling in the pathogenesis of SCLC and failure of imatinib [32][33]. Therefore, the development of an ADC would be a reasonable option to effectively treat SCLC. One important factor to consider when creating an ADC is the internalization efficiency after the complex formation of the antibody with the target molecule. Therefore, researchers first investigated whether the 4C9 antibody is internalized in SCLC cell lines. FACS analysis exhibited that the internalization efficiency of the 4C9 antibody was 91% in NCI-H526, 76.6% in NCI-H1048, and 68.6% in NCI-H889 cells (Figure 3A), which suggests that the 4C9 antibody could be used as an efficient carrier for the specific delivery of toxin to treat SCLC. Next, researchers generated an ADC using SMCC-DM1, which consists of a noncleavable linker and a microtubule inhibitor. Because DM1 absorbs ultraviolet light at 252 nm [35], the absorbance of the naked antibody (4C9) and ADC (4C9-DM1) at 252 nm was analyzed. The absorbance of 4C9-DM1 was higher than that of 4C9 at 252 nm (Figure 3B). The drug-antibody ratio (DAR) was determined as described previously [36] and the DAR was approximately 2.16. SDS-PAGE analysis exhibited that the conjugation of SMCC-DM1 to the 4C9 antibody resulted in a slight size shift. Since N-hydroxysuccinimide ester of the SMCC linker reacts with primary amines of lysine residues in the antibody, the target binding affinity of ADC may be affected. An ELISA demonstrated that the binding affinities of 4C9 and 4C9-DM1 to c-Kit were similar (Figure 3C). A quantitative analysis of the binding affinity using surface plasmon resonance (SPR) indicated that the binding affinity of 4C9 to human c-Kit was 5.5 × 10−9 M (Ka = 2.27 × 104 M−1s−1 and Kd = 1.27 × 10−4 s−1), and 4C9-DM1 also showed similar binding affinity (5.46 × 10−9 M; Ka = 2.14 × 104 M−1s−1 and Kd = 1.16 × 10−4 s−1) (Figure 3D). Taken together, these results indicate that conjugation using SMCC-DM1 to the 4C9 antibody did not affect the binding affinity of 4C9 antibody for c-Kit.
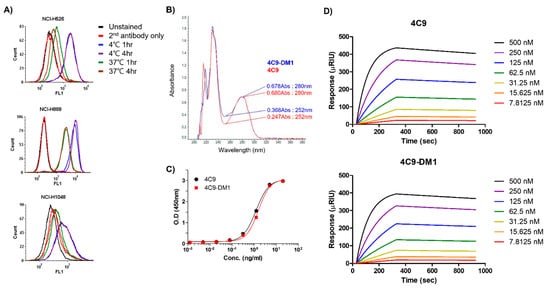
Figure 3. Characterization of 4C9-DM1. (A) The internalization of the 4C9 antibody into various SCLC cell lines was determined using FACS analysis. SCLC cells were incubated with cycloheximide (75 μg/mL) and blocked with Fc blocker for 10 min to inhibit Fc receptor-mediated internalization. SCLC cells were incubated in the presence or absence of the 4C9 antibody at 4 °C or 37 °C for 1–4 h and subjected to flow cytometry. The fluorescent signal of the 4C9/c-Kit complex on the cell surface decreased after incubation at 37 °C. (B) Optical absorbance of 4C9-DM1 compared with that of the naked 4C9 antibody at 252 nm. The binding affinity of 4C9 and 4C9-DM1 to c-Kit was compared using ELISA (C) and SPR (D).
4. 4C9-DM1 Exhibits Antitumor Activity In Vitro and In Vivo
Next, researchers analyzed the in vitro cytotoxicity using various SCLC cell lines (NCI-H526, NCI-H889, NCI-H1048, NCI-H446, and NCI-H2170) and a breast cancer cell line (MDA-MB-453). The c-Kit negative cell lines (NCI-H446, NCI-H2170, and MDA-MB-453) were used as negative controls. 4C9-DM1 exhibited in vitro cytotoxicity against NCI-H526, NCI-H889, and NCI-H1048 with half-maximal inhibitory concentration (IC50) values ranging from 158 pM to 4 nM. The in vitro cytotoxic activity of 4C9-DM1 against c-Kit-positive cancer cell lines was 4- to >300-fold higher than that against c-Kit-negative cancer cell lines (Figure 4A,B and Table 1). Interestingly, the cytotoxic activity of DM1 against the c-Kit-positive cancer cells was 7- to >77 fold higher when applied as ADC rather than as payload alone. By contrast, its cytotoxic activity was 2–5 times lower against c-Kit-negative cancer cells (Table 1), suggesting the inclusion of a payload as an ADC may reduce off-target toxicity. DM1 induces cell cycle arrest at the G2/M phase by inhibiting the assembly of microtubules, resulting in apoptosis of actively dividing cells. Therefore, researchers analyzed the effect of 4C9-DM1 on the cell cycle using NCI-H526, a c-Kit positive SCLC cell line, and NCI-H446, a c-Kit negative SCLC cell line. As shown in Figure 4C, 4C9-DM1 significantly increased the cell population in the G2/M phase in a time-dependent manner but 4C9 and IgG-DM1 did not. In addition, 4C9, 4C9-DM1, and IgG-DM1 did not alter the cell population in the G2/M phase in c-Kit negative NCI-H446 cells, suggesting that cell cycle arrest in NCI-H526 cells is specifically mediated by DM1 delivered by complex formation with 4C9-DM1 and c-Kit.
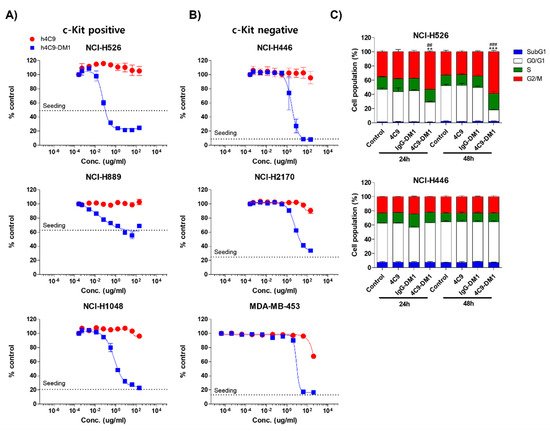
Figure 4. 4C9-DM1 exhibits cytotoxicity against SCLC cells in vitro. (A,B) c-Kit positive or negative SCLC cell lines were seeded into 96-well plates and incubated with 4C9 or 4C9-DM1 in a dose-dependent manner for 3–5 days. Live cells were stained with Hoechst 33342 (10 μM) at 37 °C for 30 min and quantitated using a Celigo Imaging Cytometer. Treatment with 4C9-DM1 decreased cell viability in a dose-dependent manner. The results represent the mean ± standard error of the mean of at least three independent experiments. (C) 4C9-DM1 induced cell cycle arrest at the G2/M phase. SCLC cell lines were seeded into 96-well plates and incubated with 4C9 (1 μg/mL), IgG-DM1 (1 μg/mL), or 4C9-DM1 (1 μg/mL) for 24 and 48 h. Then, the cells were stained with propidium iodide and analyzed using a Celigo Imaging Cytometer (** and *** vs. control; ## and ### vs. IgG-DM1). The results represent the mean ± standard error of the mean of at least three independent experiments. The results are presented as the mean ± standard error of the mean. The means were compared using an unpaired Student’s two-sided t-test. ** p < 0.01, *** p < 0.001, ## p < 0.01, ### p < 0.001.
Table 1. IC50 (nM) values of the tested materials.
| c-Kit Expression | Tissue Type | Cell Line | 4C9-DM1 * | SMCC-DM1 |
|---|---|---|---|---|
| c-Kit positive | SCLC | NCI-H526 | 0.158 | 12.23 |
| NCI-H889 | 0.323 | 11.62 | ||
| NCI-H1048 | 4.08 | 30.45 | ||
| c-Kit negative | SCLC | NCI-H446 | 16.58 | 6.97 |
| NCI-H2170 | 35.5 | 20.17 | ||
| Breast cancer | MDA-MB-453 | 47.63 | 9.763 |
* Molar concentration was calculated as 180 kDa for the molecular weight of 4C9-DM1.
Based on the in vitro cytotoxicity assay, the in vivo efficacy of 4C9-DM1 was examined using mouse models xenotransplanted with NCI-H526. Although IgG-DM1 did not exert an effect, 4C9-DM1 significantly suppressed NCI-H526 tumor growth in a dose-dependent manner (Figure 5A). Tumor growth inhibition (TGI) rates of 4C9-DM1 at doses of 1, 3, and 5 mg/kg were 40%, 45%, and 59%, respectively, compared with that of the vehicle control. The 4C9 antibody alone partially inhibited tumor growth, but this effect was not statistically significant. Body weight losses due to the administered materials were not observed (Figure 5A), suggesting that there was no concern related to toxicity. Chemotherapy, including etoposide, cisplatin, carboplatin, and lurbinectedin are used to treat SCLC patients as the standard of care [37][38]. Although combination therapy using chemotherapeutic drugs is effective, most SCLCs rapidly recur within 1 year [39]. Therefore, researchers determined whether the combination of 4C9-DM1 with chemotherapy could enhance the therapeutic efficacy of SCLC. As shown in Figure 5B, 4C9-DM1, lurbinectedin, and carboplatin/etoposide exhibited similar antitumor activities; the TGI rates of these groups were 50% at day 18, compared with that of the vehicle control. Interestingly, the combination of 4C9-DM1 with lurbinectedin or carboplatin/etoposide synergistically suppressed tumor growth. The TGI rates of both groups were 85%, compared with that of the vehicle control at day 18 with subsequent regrowth. The combinatorial treatment with 4C9-DM1 plus lurbinectedin induced body weight loss of approximately 10%; however, body weight increased again after cessation of the treatment (Figure 5B). This suggests that this combination may be used to treat SCLC with manageable or mild toxicity.
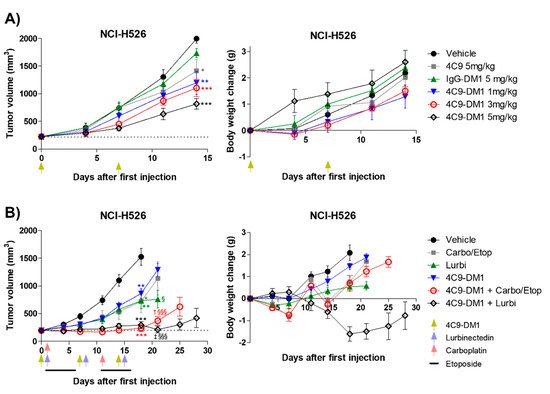
Figure 5. 4C9−DM1 suppresses SCLC tumor growth in a xenograft mouse model. (A,B) Antitumor activity of 4C9-DM1 was evaluated in an in vivo xenograft mouse model. NCI-H526 cancer cells were implanted into immune-deficient mice as described in the Methods section. Mice with established tumors were randomized into different treatment groups when the tumor volume reached ~200 mm3 (n = 6). The animals were intravenously administered vehicle, 4C9, IgG-DM1, or 4C9-DM1. Carboplatin (60 mg/kg on days 1 and 11) and etoposide (3 mg/kg on days 1–5 and days 11–15) were intraperitoneally administered or combined with 4C9-DM1. Additionally, lurbinectedin (0.08 mg/kg on days 1, 8, and 15) was intravenously administered or combined with 4C9-DM1 as indicated. Green arrows indicate the administration of vehicle, IgG-DM1, 4C9, or 4C9-DM1, and blue and red arrows indicate the administration of lurbinectedin and carboplatin, respectively (*, **, and *** vs. their respective corresponding vehicle control; § and §§§ vs. their respective corresponding 4C9-DM1 control; † vs. carboplatin/etoposide; ‡ vs. lurbinectedin). The results are presented as the mean ± standard error of the mean. The means were compared using an unpaired Student’s two-sided t-test. * p < 0.05, ** p < 0.01, *** p < 0.001, † p < 0.05, ‡ p < 0.01, § p < 0.05, §§§ p < 0.001.
References
- Blandin Knight, S.; Crosbie, P.A.; Balata, H.; Chudziak, J.; Hussell, T.; Dive, C. Progress and prospects of early detection in lung cancer. Open Biol. 2017, 7, 170070.
- Elias, A.D. Small cell lung cancer: State-of-the-art therapy in 1996. Chest 1997, 112, 251S–258S.
- Dores, G.M.; Qubaiah, O.; Mody, A.; Ghabach, B.; Devesa, S.S. A population-based study of incidence and patient survival of small cell carcinoma in the United States, 1992–2010. BMC Cancer 2015, 15, 185.
- Wang, S.; Tang, J.; Sun, T.; Zheng, X.; Li, J.; Sun, H.; Zhou, X.; Zhou, C.; Zhang, H.; Cheng, Z.; et al. Survival changes in patients with small cell lung cancer and disparities between different sexes, socioeconomic statuses and ages. Sci. Rep. 2017, 7, 1339.
- Salgia, R.; Skarin, A.T. Molecular abnormalities in lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1998, 16, 1207–1217.
- Rozengurt, E. Autocrine loops, signal transduction, and cell cycle abnormalities in the molecular biology of lung cancer. Curr. Opin. Oncol. 1999, 11, 116–122.
- Wojtalla, A.; Fischer, B.; Kotelevets, N.; Mauri, F.A.; Sobek, J.; Rehrauer, H.; Wotzkow, C.; Tschan, M.P.; Seckl, M.J.; Zangemeister-Wittke, U.; et al. Targeting the phosphoinositide 3-kinase p110-alpha isoform impairs cell proliferation, survival, and tumor growth in small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 96–105.
- Kim, Y.H.; Girard, L.; Giacomini, C.P.; Wang, P.; Hernandez-Boussard, T.; Tibshirani, R.; Minna, J.D.; Pollack, J.R. Combined microarray analysis of small cell lung cancer reveals altered apoptotic balance and distinct expression signatures of MYC family gene amplification. Oncogene 2006, 25, 130–138.
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretic, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53.
- Carvajal, L.A.; Manfredi, J.J. Another fork in the road--life or death decisions by the tumour suppressor p53. EMBO Rep. 2013, 14, 414–421.
- Gazdar, A.F.; Bunn, P.A.; Minna, J.D. Small-cell lung cancer: What we know, what we need to know and the path forward. Nat. Rev. Cancer 2017, 17, 765.
- Carter, B.W.; Glisson, B.S.; Truong, M.T.; Erasmus, J.J. Small cell lung carcinoma: Staging, imaging, and treatment considerations. Radiographics 2014, 34, 1707–1721.
- Pleasance, E.D.; Stephens, P.J.; O’Meara, S.; McBride, D.J.; Meynert, A.; Jones, D.; Lin, M.L.; Beare, D.; Lau, K.W.; Greenman, C.; et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 2010, 463, 184–190.
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622.
- Yarden, Y.; Kuang, W.J.; Yang-Feng, T.; Coussens, L.; Munemitsu, S.; Dull, T.J.; Chen, E.; Schlessinger, J.; Francke, U.; Ullrich, A. Human proto-oncogene c-kit: A new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987, 6, 3341–3351.
- Geissler, E.N.; Ryan, M.A.; Housman, D.E. The dominant-white spotting (W) locus of the mouse encodes the c-kit proto-oncogene. Cell 1988, 55, 185–192.
- Ashman, L.K. The biology of stem cell factor and its receptor C-kit. Int. J. Biochem. Cell Biol. 1999, 31, 1037–1051.
- Vose, J.M.; Armitage, J.O. Clinical applications of hematopoietic growth factors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1995, 13, 1023–1035.
- Lennartsson, J.; Ronnstrand, L. Stem cell factor receptor/c-Kit: From basic science to clinical implications. Physiol. Rev. 2012, 92, 1619–1649.
- Bougherara, H.; Subra, F.; Crepin, R.; Tauc, P.; Auclair, C.; Poul, M.A. The aberrant localization of oncogenic kit tyrosine kinase receptor mutants is reversed on specific inhibitory treatment. Mol. Cancer Res. MCR 2009, 7, 1525–1533.
- Krystal, G.W.; Hines, S.J.; Organ, C.P. Autocrine growth of small cell lung cancer mediated by coexpression of c-kit and stem cell factor. Cancer Res. 1996, 56, 370–376.
- Hibi, K.; Takahashi, T.; Sekido, Y.; Ueda, R.; Hida, T.; Ariyoshi, Y.; Takagi, H.; Takahashi, T. Coexpression of the stem cell factor and the c-kit genes in small-cell lung cancer. Oncogene 1991, 6, 2291–2296.
- Sekido, Y.; Obata, Y.; Ueda, R.; Hida, T.; Suyama, M.; Shimokata, K.; Ariyoshi, Y.; Takahashi, T. Preferential expression of c-kit protooncogene transcripts in small cell lung cancer. Cancer Res. 1991, 51, 2416–2419.
- Hida, T.; Ueda, R.; Sekido, Y.; Hibi, K.; Matsuda, R.; Ariyoshi, Y.; Sugiura, T.; Takahashi, T.; Takahashi, T. Ectopic expression of c-kit in small-cell lung cancer. Int. J. Cancer. Suppl. = J. Int. Du Cancer. Suppl. 1994, 8, 108–109.
- Zhu, Y.; Qian, S.X. Clinical efficacy and safety of imatinib in the management of Ph(+) chronic myeloid or acute lymphoblastic leukemia in Chinese patients. OncoTargets Ther. 2014, 7, 395–404.
- Lopes, L.F.; Bacchi, C.E. Imatinib treatment for gastrointestinal stromal tumour (GIST). J. Cell. Mol. Med. 2010, 14, 42–50.
- Helbig, G. Imatinib for the treatment of hypereosinophilic syndromes. Expert Rev. Clin. Immunol. 2018, 14, 163–170.
- Sacha, T. Imatinib in chronic myeloid leukemia: An overview. Mediterr. J. Hematol. Infect. Dis. 2014, 6, e2014007.
- Krystal, G.W.; Honsawek, S.; Litz, J.; Buchdunger, E. The selective tyrosine kinase inhibitor STI571 inhibits small cell lung cancer growth. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 3319–3326.
- Wang, W.L.; Healy, M.E.; Sattler, M.; Verma, S.; Lin, J.; Maulik, G.; Stiles, C.D.; Griffin, J.D.; Johnson, B.E.; Salgia, R. Growth inhibition and modulation of kinase pathways of small cell lung cancer cell lines by the novel tyrosine kinase inhibitor STI 571. Oncogene 2000, 19, 3521–3528.
- Soria, J.C.; Johnson, B.E.; Chevalier, T.L. Imatinib in small cell lung cancer. Lung Cancer 2003, 41 (Suppl. S1), S49–S53.
- Johnson, B.E.; Fischer, T.; Fischer, B.; Dunlop, D.; Rischin, D.; Silberman, S.; Kowalski, M.O.; Sayles, D.; Dimitrijevic, S.; Fletcher, C.; et al. Phase II study of imatinib in patients with small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 5880–5887.
- Dy, G.K.; Miller, A.A.; Mandrekar, S.J.; Aubry, M.C.; Langdon, R.M., Jr.; Morton, R.F.; Schild, S.E.; Jett, J.R.; Adjei, A.A. A phase II trial of imatinib (ST1571) in patients with c-kit expressing relapsed small-cell lung cancer: A CALGB and NCCTG study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2005, 16, 1811–1816.
- Kim, J.O.; Kim, K.H.; Baek, E.J.; Park, B.; So, M.K.; Ko, B.J.; Ko, H.J.; Park, S.G. A novel anti-c-Kit antibody-drug conjugate to treat wild-type and activating-mutant c-Kit-positive tumors. Mol. Oncol. 2021.
- Wagh, A.; Song, H.; Zeng, M.; Tao, L.; Das, T.K. Challenges and new frontiers in analytical characterization of antibody-drug conjugates. mAbs 2018, 10, 222–243.
- Tedeschini, T.; Campara, B.; Grigoletto, A.; Bellini, M.; Salvalaio, M.; Matsuno, Y.; Suzuki, A.; Yoshioka, H.; Pasut, G. Polyethylene glycol-based linkers as hydrophilicity reservoir for antibody-drug conjugates. J. Control. Release Off. J. Control. Release Soc. 2021, 337, 431–447.
- Singh, S.; Jaigirdar, A.A.; Mulkey, F.; Cheng, J.; Hamed, S.S.; Li, Y.; Liu, J.; Zhao, H.; Goheer, A.; Helms, W.S.; et al. FDA Approval Summary: Lurbinectedin for the Treatment of Metastatic Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 2378–2382.
- Farago, A.F.; Keane, F.K. Current standards for clinical management of small cell lung cancer. Transl. Lung Cancer Res. 2018, 7, 69–79.
- Gong, J.; Salgia, R. Managing Patients With Relapsed Small-Cell Lung Cancer. J. Oncol. Pract. 2018, 14, 359–366.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
3 times
(View History)
Update Date:
29 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No