 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Stefano Perni | + 3211 word(s) | 3211 | 2022-01-20 10:52:11 | | | |
| 2 | Lindsay Dong | Meta information modification | 3211 | 2022-03-01 03:06:15 | | | | |
| 3 | Lindsay Dong | Meta information modification | 3211 | 2022-03-01 03:07:26 | | |
Video Upload Options
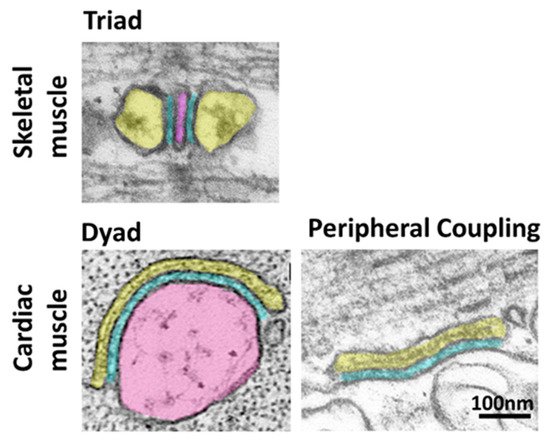
Contraction of striated muscle is triggered by a massive release of calcium from the sarcoplasmic reticulum (SR) into the cytoplasm. This intracellular calcium release is initiated by membrane depolarization, which is sensed by voltage-gated calcium channels CaV1.1 (in skeletal muscle) and CaV1.2 (in cardiac muscle) in the plasma membrane (PM), which in turn activate the calcium-releasing channel ryanodine receptor (RyR) embedded in the SR membrane. This cross-communication between channels in the PM and in the SR happens at specialized regions, the SR-PM junctions, where these two compartments come in close proximity. Junctophilin1 and Junctophilin2 are responsible for the formation and stabilization of SR-PM junctions in striated muscle and actively participate in the recruitment of the two essential players in intracellular calcium release, CaV and RyR.
1. Introduction
2. The Junctophilin Family
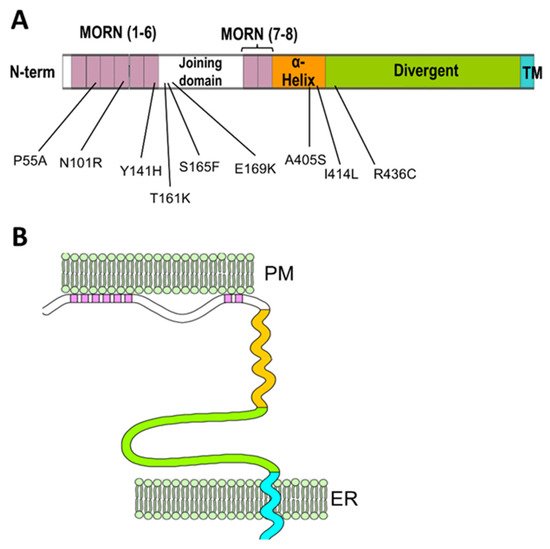
3. Recruitment of Junctional Proteins by Junctophilins1 and 2
3.1. Recruitment of Skeletal Muscle Junctional Proteins
3.2. Recruitment of Cardiac Muscle Junctional Proteins
4. Functional Studies on Junctophilins 1 and 2
4.1. Junctophilin 1
JPH1 knock-out mice die within 24 h after birth due to suckling defects leading to undernourishment. The suckling defect is likely due to muscle weakness since the neuronal suckling reflexes are normal in knock-out mice [37]. Functional studies on isolated hindlimb muscle showed abnormal twitch tension and a greater dependency on extracellular calcium in KO mice muscles, suggesting that a significant fraction of RyR1s in the junctional SR are not directly coupled with the CaV1.1 channels in the T-tubules and therefore operate via calcium-induced calcium release. Nonetheless, knock-out (KO) mice are still relatively mobile and show skeletal muscle-type EC coupling to a certain degree, indicating that JPH2 can support voltage-induced Ca2+ release in the absence of JPH1. From a structural point of view, although no major disorganization of the fiber is noticed at the light microscopy level, evident alterations are noticeable at the ultrastructural level [31][38]. In particular, the skeletal muscle of wt and JPH1 KO mice show a similar development in the embryonic stages until shortly after birth. At this age, wt muscle experiences a significant increase in JPH1 expression, which is temporally correlated with the transition from immature SR-PM junctions, mainly organized in dyads at this stage, into fully formed triads. This transition is absent in JPH1 KO muscle [31][38], suggesting that JPH2 is important in forming the dyads, while JPH1 has a crucial role in the conversion from dyads to triads in the fully mature skeletal muscle. The knocking down of junctophilins using sh-RNA, leads to the impairment of store-operated Ca2+ entry (SOCE), altered intracellular calcium release and intracellular calcium stores [39] and to a reduction in RyR1 and CaV1.1 co-clustering associated with a decrease in CaV1.1 membrane expression [27] both in myotubes and muscle fibers. In both these studies, a shRNA against both JPH1 and JPH2 was used; hence, it was impossible to distinguish each isoform’s relative contribution to the resulting phenotype.
4.2. Junctophilin 2
5. Post-Transcriptional Regulation of JPH1 and JPH2
6. New Insights from Deep Learning Protein Structure Prediction
Remarkable advancements in protein folding prediction were recently achieved by the artificial intelligence software Alphafold2 [47]. Alphafold2 is a giant leap forward in the reliability of protein folding prediction compared to similar existing software [48], and it has already been used to predict the structure of nearly the entire human proteome. Based on Aplhafold2 prediction models, junctophilin 1 and 2 show a similar 3D structure, also shared by the neuronal isoforms JPH3 and JPH4 (UniProt protein ID: Q9HDC5, Q9BR39, Q8WXH2, Q96JJ6 for human JPH1, JPH2, JPH3 and JPH4, respectively). The structure of the most ordered domains of junctophilin, specifically the MORN repeats, the α-helical region and the transmembrane domain, are predicted with high confidence by Alphafold2. In contrast, the joining and divergent domains are likely disordered, at least in the isolated protein, and the structure cannot be predicted with reasonable confidence. According to the prediction model (Figure 3), the MORN domains are arranged in an extended “halfpipe” configuration, with the α-helical domain lying on the convex side of this half-pipe (Figure 3B,C) and establishing interactions with charged residues in the MORN domains (see the zoomed-in region in Figure 3C for an example).
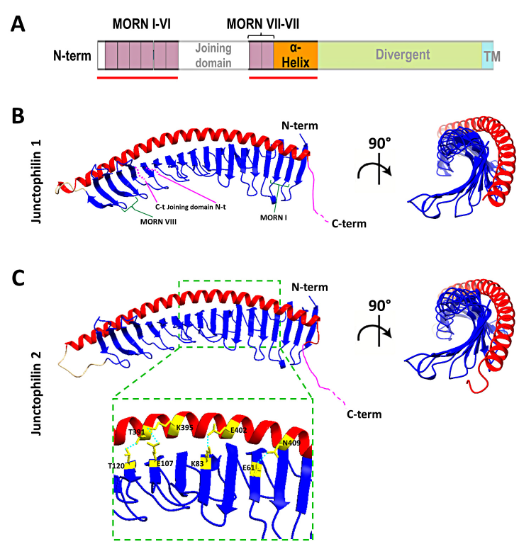
Figure 3. Structure of the MORNs and α-helical domains of human junctophilin1 and junctophilin2.(A) Schematic representation of junctophilin domain as shown in Figure 2; the solid red lines indicate the regions for which the structure is predicted with high fidelity by Alphafold2 and illustrated in (B,C). (B,C) predicted structures of the MORNs- -helical domains of junctophilin1 (B) and junctophilin2 (C). The α-helical domain (in red) lies on the convex side of the MORN domains half-pipe structure (in blue) in both junctophilin1 and junctophilin2. β-sheet hairpins forming MORN domains I and VIII (green parentheses) and the position of the N-terminal end (N-t) and C-terminal end (C-t) of the joining domain (in pink), which is absent in this representation, are indicated in (B). The inset in (C) shows some of the residues that form the hydrogen bonds that stabilize the association between the MORN domains and the α-helical domain of junctophilin2.
The structure obtained using Alphafold2 is substantially different from what was previously predicted using RaptorX software by Gross and colleagues [49]. In the structure described by Gross et al., the α-helical domain extends beyond the MORN domains without interacting with them at all. However, Alphafold2 software is considered to be more accurate than most (if not all) of the currently existing structure-predicting software, especially for proteins for which no homologous structures exist [50][51], and the reciprocal arrangement of JPH2 MORN motifs and α-helical domain predicted by Alphafold2 agrees with data from Li and collaborators [52] based on the crystal structure of the protein MORN4. MORN4 contains a series of MORN motifs arranged in a half-pipe configuration followed by a brief α-helical region. The helical region stabilizes the MORN domains by lying over part of the convex side of the half-pipe. The structure solved by Li and colleagues is in many ways very similar to the sequence predicted by Alphafold2 for JPHs.
Furthermore, in MORN 4, the concave side of the MORN half-pipe structure, containing most of the conserved residues that define the MORN domain, engages in the binding with the α-helical region of myosin3a. It is conceivable that the concave side of the junctophilin MORN motifs could also participate in protein–protein interactions with components of the EC coupling machinery. The particular arrangement of the α-helical domain with respect to the MORN motifs predicted by Alphafold2 and suggested by the observations of Li and colleagues challenges the classic view of the α-helical domain as the spacer that spans most of the junctional gap (see schematic representation in Figure 2) and points to the divergent domain as the region that most likely fulfills this role.
References
- A. F. Huxley; R. E. Taylor; Local activation of striated muscle fibres. The Journal of Physiology 1958, 144, 426-441, 10.1113/jphysiol.1958.sp006111.
- A F Huxley; Local activation of striated muscle from the frog and the crab.. The Journal of Physiology 1957, 135, 269-300.
- Keith R. Porter; George E. Palade; STUDIES ON THE ENDOPLASMIC RETICULUM. Journal of Cell Biology 1957, 3, 269-300, 10.1083/jcb.3.2.269.
- C Franzini-Armstrong; FINE STRUCTURE OF SARCOPLASMIC RETICULUM AND TRANVERSE TUBULAR SYSTEM IN MUSCLE FIBERS.. Federation proceedings 1964, 23, 887-895.
- Clara Franzini-Armstrong; STUDIES OF THE TRIAD. Journal of Cell Biology 1970, 47, 488-499, 10.1083/jcb.47.2.488.
- Matthew T. Close; Stefano Perni; Clara Franzini-Armstrong; David Cundall; Highly extensible skeletal muscle in snakes. Journal of Experimental Biology 2013, 217, 2445-8, 10.1242/jeb.097634.
- L. C. Rome; D. A. Syme; S. Hollingworth; S. L. Lindstedt; S. M. Baylor; The whistle and the rattle: the design of sound producing muscles.. Proceedings of the National Academy of Sciences 1996, 93, 8095-8100, 10.1073/pnas.93.15.8095.
- Clara Franzini-Armstrong; Feliciano Protasi; Venkat Ramesh; Shape, Size, and Distribution of Ca2+ Release Units and Couplons in Skeletal and Cardiac Muscles. Biophysical Journal 1999, 77, 1528-1539, 10.1016/s0006-3495(99)77000-1.
- Stefano Perni; Kurt C. Marsden; Matias Escobar; Stephen Hollingworth; Stephen M. Baylor; Clara Franzini-Armstrong; Structural and functional properties of ryanodine receptor type 3 in zebrafish tail muscle. Journal of General Physiology 2015, 145, 173-184, 10.1085/jgp.201411303.
- Manuela Lavorato; Tai-Qin Huang; Venkat Ramesh Iyer; Stefano Perni; Gerhard Meissner; Clara Franzini-Armstrong; Dyad content is reduced in cardiac myocytes of mice with impaired calmodulin regulation of RyR2. Journal of Muscle Research and Cell Motility 2015, 36, 205-214, 10.1007/s10974-015-9405-5.
- Stefano Perni; V. Ramesh Iyer; Clara Franzini-Armstrong; Ultrastructure of cardiac muscle in reptiles and birds: optimizing and/or reducing the probability of transmission between calcium release units. Journal of Muscle Research and Cell Motility 2012, 33, 145-152, 10.1007/s10974-012-9297-6.
- A Fabiato; Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell.. Journal of General Physiology 1985, 85, 247-289, 10.1085/jgp.85.2.247.
- Barbara A Block; Toshiaki Imagawa; Kevin Campbell; Clara Franziniarmstrong; Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle.. Journal of Cell Biology 1988, 107, 2587-2600, 10.1083/jcb.107.6.2587.
- Johann Schredelseker; Anamika Dayal; Thorsten Schwerte; Clara Franzini-Armstrong; Manfred Grabner; Proper Restoration of Excitation-Contraction Coupling in the Dihydropyridine Receptor β1-null Zebrafish Relaxed Is an Exclusive Function of the β1a Subunit. Journal of Biological Chemistry 2008, 284, 1242-1251, 10.1074/jbc.m807767200.
- Petra Weissgerber; Brigitte Held; Wilhelm Bloch; Lars Kaestner; Kenneth R. Chien; Bernd K. Fleischmann; Peter Lipp; Veit Flockerzi; Marc Freichel; Reduced Cardiac L-Type Ca 2+ Current in Ca v β 2 −/− Embryos Impairs Cardiac Development and Contraction With Secondary Defects in Vascular Maturation. Circulation Research 2006, 99, 749-757, 10.1161/01.res.0000243978.15182.c1.
- Marion Pragnell; Michel De Waard; Yasuo Mori; Tsutomu Tanabe; Terry P. Snutch; Kevin Campbell; Calcium channel β-subunit binds to a conserved motif in the I–II cytoplasmic linker of the α1-subunit. Nature 1994, 368, 67-70, 10.1038/368067a0.
- Andy J. Chien; Xiaolan Zhao; Roman E. Shirokov; Tipu S. Puri; Chan Fong Chang; Dandan Sun; Eduardo Rios; M. Marlene Hosey; Roles of a Membrane-localized βSubunit in the Formation and Targeting of Functional L-type Ca2+ Channels. Journal of Biological Chemistry 1995, 270, 30036-30044, 10.1074/jbc.270.50.30036.
- Benjamin R. Nelson; Fenfen Wu; Yun Liu; Douglas M. Anderson; John McAnally; Weichun Lin; Stephen Cannon; Rhonda Bassel-Duby; Eric N. Olson; Skeletal muscle-specific T-tubule protein STAC3 mediates voltage-induced Ca2+ release and contractility. Proceedings of the National Academy of Sciences 2013, 110, 11881-11886, 10.1073/pnas.1310571110.
- Eric Horstick; Jeremy Linsley; James J. Dowling; Michael A. Hauser; Kristin K. McDonald; Allison Ashley-Koch; Louis Saint-Amant; Akhila Satish; Wilson W. Cui; Weibin Zhou; et al.Shawn M. SpragueDemetra S. StammCynthia M. PowellMarcy C. SpeerClara Franzini-ArmstrongHiromi HirataJohn Y. Kuwada Stac3 is a component of the excitation–contraction coupling machinery and mutated in Native American myopathy. Nature Communications 2013, 4, 1-11, 10.1038/ncomms2952.
- Alexander Polster; Stefano Perni; Hicham Bichraoui; Kurt G. Beam; Stac adaptor proteins regulate trafficking and function of muscle and neuronal L-type Ca2+ channels. Proceedings of the National Academy of Sciences 2014, 112, 602-606, 10.1073/pnas.1423113112.
- Alexander Polster; Benjamin R. Nelson; Eric N. Olson; Kurt G. Beam; Stac3 has a direct role in skeletal muscle-type excitation–contraction coupling that is disrupted by a myopathy-causing mutation. Proceedings of the National Academy of Sciences 2016, 113, 10986-10991, 10.1073/pnas.1612441113.
- Alexander Polster; Benjamin R. Nelson; Symeon Papadopoulos; Eric N. Olson; Kurt G. Beam; Stac proteins associate with the critical domain for excitation–contraction coupling in the II–III loop of CaV1.1. Journal of General Physiology 2018, 150, 613-624, 10.1085/jgp.201711917.
- Tsutomu Tanabe; Kurt G. Beam; Brett A. Adams; Tetsuhiro Niidome; Shosaku Numa; Regions of the skeletal muscle dihydropyridine receptor critical for excitation–contraction coupling. Nature 1990, 346, 567-569, 10.1038/346567a0.
- Miyuki Nishi; Hiroyuki Sakagami; Shinji Komazaki; Hisatake Kondo; Hiroshi Takeshima; Coexpression of junctophilin type 3 and type 4 in brain. Molecular Brain Research 2003, 118, 102-110, 10.1016/s0169-328x(03)00341-3.
- H Takeshima; JunctophilinsA Novel Family of Junctional Membrane Complex Proteins. Molecular Cell 2000, 6, 11-22, 10.1016/s1097-2765(00)00003-4.
- Alejandro Garbino; Ralph J. Van Oort; Sayali S. Dixit; Andrew P. Landstrom; Michael J. Ackerman; Xander H. T. Wehrens; Molecular evolution of the junctophilin gene family. Physiological Genomics 2009, 37, 175-186, 10.1152/physiolgenomics.00017.2009.
- Lucia Golini; Christophe Chouabe; Christine Berthier; Vincenza Cusimano; Mara Fornaro; Robert Bonvallet; Luca Formoso; Emiliana Giacomello; Vincent Jacquemond; Vincenzo Sorrentino; et al. Junctophilin 1 and 2 Proteins Interact with the L-type Ca2+ Channel Dihydropyridine Receptors (DHPRs) in Skeletal Muscle. Journal of Biological Chemistry 2011, 286, 43717-43725, 10.1074/jbc.m111.292755.
- Tsutomu Nakada; Toshihide Kashihara; Masatoshi Komatsu; Katsuhiko Kojima; Toshikazu Takeshita; Mitsuhiko Yamada; Physical interaction of junctophilin and the CaV1.1 C terminus is crucial for skeletal muscle contraction. Proceedings of the National Academy of Sciences 2018, 115, 4507-4512, 10.1073/pnas.1716649115.
- Stefano Perni; Manuela Lavorato; Kurt G. Beam; De novo reconstitution reveals the proteins required for skeletal muscle voltage-induced Ca2+ release. Proceedings of the National Academy of Sciences 2017, 114, 13822-13827, 10.1073/pnas.1716461115.
- Phimister, A.J.; Lango, J.; Lee, E.H.; Ernst-Russell, M.A.; Takeshima, H.; Ma, J.; Allen, P.D.; Pessah, I.N. Conformation-dependent stability of junctophilin 1 (JP1) and ryanodine receptor type 1 (RyR1) channel complex is mediated by their hyper-reactive thiols. J. Biol. Chem. 2007, 282, 8667–8677.
- Koichi Ito; Shinji Komazaki; Kazushige Sasamoto; Morikatsu Yoshida; Miyuki Nishi; Kenji Kitamura; Hiroshi Takeshima; Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. Journal of Cell Biology 2001, 154, 1059-1068, 10.1083/jcb.200105040.
- Stefano Perni; Kurt Beam; Junctophilins 1, 2, and 3 all support voltage-induced Ca2+ release despite considerable divergence. Journal of General Physiology 2022, 154, e202113024, 10.1085/jgp.202113024.
- Min Jiang; Mei Zhang; Maureen Howren; Yuhong Wang; Alex Tan; Ravi C. Balijepalli; Jose Huizar; Gea-Ny Tseng; JPH-2 interacts with Cai-handling proteins and ion channels in dyads: Contribution to premature ventricular contraction–induced cardiomyopathy. Heart Rhythm 2015, 13, 743-752, 10.1016/j.hrthm.2015.10.037.
- Ang Guo; Duane Hall; Caimei Zhang; Tianqing Peng; Jordan Miller; William Kutschke; Chad E. Grueter; Frances L. Johnson; Richard Lin; Long-Sheng Song; et al. Molecular Determinants of Calpain-dependent Cleavage of Junctophilin-2 Protein in Cardiomyocytes. Journal of Biological Chemistry 2015, 290, 17946-17955, 10.1074/jbc.m115.652396.
- David L. Beavers; Wei Wang; Sameer Ather; Niels Voigt; Alejandro Garbino; Sayali S. Dixit; Andrew Landstrom; Na Li; Qiongling Wang; Iacopo Olivotto; et al.Dobromir DobrevMichael J. AckermanXander H.T. Wehrens Mutation E169K in Junctophilin-2 Causes Atrial Fibrillation Due to Impaired RyR2 Stabilization. Journal of the American College of Cardiology 2013, 62, 2010-2019, 10.1016/j.jacc.2013.06.052.
- Liheng Yin; Alexandra Zahradnikova Jr; Riccardo Rizzetto; Simona Boncompagni; Camille Rabesahala de Meritens; Yadan Zhang; Pierre Joanne; Elena Marques Sule; Yuriana Aguilar-Torres; Miguel Fernandez-Tenorio; et al.Olivier VillejoubertLinwei LiYue Yi WangPhilippe MateoValerie NicolasPascale GerbaudF Anthony LaiRomain PerrierJulio L AlvarezErnst NiggliHéctor H ValdiviaCarmen R ValdiviaJosefina Ramos-FrancoEsther ZorioSpyros ZissimopoulosFeliciano ProtasiJean-Pierre BenitahAna-Maria Gomez Impaired Binding to Junctophilin2 and Nanostructural Alteration in CPVT Mutation. Circulation Research 2021, 129, e35-e52, 10.1161/circresaha.121.319094.
- Ito, K.; Komazaki, S.; Sasamoto, K.; Yoshida, M.; Nishi, M.; Kitamura, K.; Takeshima, H. Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J. Cell Biol. 2001, 154, 1059–1067.
- Shinji Komazaki; Koichi Ito; Hiroshi Takeshima; Hiroaki Nakamura; Deficiency of triad formation in developing skeletal muscle cells lacking junctophilin type 1. FEBS Letters 2002, 524, 225-229, 10.1016/s0014-5793(02)03042-9.
- Yutaka Hirata; Marco Brotto; Noah Weisleder; Yi Chu; Pei-Hui Lin; Xiaoli Zhao; Angela Thornton; Shinji Komazaki; Hiroshi Takeshima; Jianjie Ma; et al.Zui Pan Uncoupling Store-Operated Ca2+ Entry and Altered Ca2+ Release from Sarcoplasmic Reticulum through Silencing of Junctophilin Genes. Biophysical Journal 2006, 90, 4418-4427, 10.1529/biophysj.105.076570.
- Ralph J. Van Oort; Alejandro Garbino; Wei Wang; Sayali S. Dixit; Andrew Landstrom; Namit Gaur; Angela C. De Almeida; Darlene G. Skapura; Yoram Rudy; Alan R. Burns; et al.Michael J. AckermanXander H. T. Wehrens Disrupted Junctional Membrane Complexes and Hyperactive Ryanodine Receptors After Acute Junctophilin Knockdown in Mice. Circulation 2011, 123, 979-988, 10.1161/circulationaha.110.006437.
- Andrew Landstrom; Noah Weisleder; Karin B. Batalden; J. Martijn Bos; David J. Tester; Steve R. Ommen; Xander Wehrens; William C. Claycomb; Jae-Kyun Ko; Moonsun Hwang; et al.Zui PanJianjie MaMichael J. Ackerman Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. Journal of Molecular and Cellular Cardiology 2007, 42, 1026-1035, 10.1016/j.yjmcc.2007.04.006.
- Jin Seok Woo; Chung-Hyun Cho; Keon Jin Lee; Do Han Kim; Jianjie Ma; Eun Hui Lee; Hypertrophy in Skeletal Myotubes Induced by Junctophilin-2 Mutant, Y141H, Involves an Increase in Store-operated Ca2+ Entry via Orai1*. Journal of Biological Chemistry 2012, 287, 14336-14348, 10.1074/jbc.m111.304808.
- R. M. Murphy; T. L. Dutka; D. Horvath; J. R. Bell; L. M. Delbridge; G. D. Lamb; Ca2+-dependent proteolysis of junctophilin-1 and junctophilin-2 in skeletal and cardiac muscle. The Journal of Physiology 2012, 591, 719-729, 10.1113/jphysiol.2012.243279.
- Eshwar R. Tammineni; Lourdes Figueroa; Natalia Kraeva; Carlo Manno; Carlos A. Ibarra; Amira Klip; Sheila Riazi; Eduardo Rios; Fragmentation and roles of junctophilin1 in muscle of patients with cytosolic leak of stored calcium. Journal of General Physiology 2021, 154, 2021ecc32, 10.1085/jgp.2021ecc32.
- Ang Guo; Yihui Wang; Biyi Chen; Yunhao Wang; Jinxiang Yuan; Liyang Zhang; Duane Hall; Jennifer Wu; Yun Shi; Qi Zhu; et al.Cheng ChenWilliam H. ThielXin ZhanRobert M. WeissFenghuang ZhanCatherine A. MusselmanMiles PufallWeizhong ZhuKin Fai AuJiang HongMark E. AndersonChad E. GrueterLong-Sheng Song E-C coupling structural protein junctophilin-2 encodes a stress-adaptive transcription regulator. Science 2018, 362, science.aan3303, 10.1126/science.aan3303.
- Chia‐Yen C. Wu; Biyi Chen; Ya‐Ping Jiang; Zhiheng Jia; Dwight W. Martin; Shengnan Liu; Emilia Entcheva; Long‐Sheng Song; Richard Z. Lin; Calpain‐Dependent Cleavage of Junctophilin‐2 and T‐Tubule Remodeling in a Mouse Model of Reversible Heart Failure. Journal of the American Heart Association 2014, 3, e000527, 10.1161/jaha.113.000527.
- John Jumper; Richard Evans; Alexander Pritzel; Tim Green; Michael Figurnov; Olaf Ronneberger; Kathryn Tunyasuvunakool; Russ Bates; Augustin Žídek; Anna Potapenko; et al.Alex BridglandClemens MeyerSimon A. A. KohlAndrew J. BallardAndrew CowieBernardino Romera-ParedesStanislav NikolovRishub JainJonas AdlerTrevor BackStig PetersenDavid ReimanEllen ClancyMichal ZielinskiMartin SteineggerMichalina PacholskaTamas BerghammerSebastian BodensteinDavid SilverOriol VinyalsAndrew W. SeniorKoray KavukcuogluPushmeet KohliDemis Hassabis Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583-589, 10.1038/s41586-021-03819-2.
- Ewen Callaway; ‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures. Nature 2020, 588, 203-204, 10.1038/d41586-020-03348-4.
- Polina Gross; Jaslyn Johnson; Carlos M. Romero; Deborah M. Eaton; Claire Poulet; Jose Sanchez-Alonso; Carla Lucarelli; Jean Ross; Andrew A. Gibb; Joanne F. Garbincius; et al.Jonathan LambertErdem VarolYijun YangMarkus WallnerEric A. FeldsottHajime KuboRemus M. BerrettaDaohai YuVictor RizzoJohn ElrodAbdelkarim SabriJulia GorelikXiongwen ChenSteven R. Houser Interaction of the Joining Region in Junctophilin-2 With the L-Type Ca 2+ Channel Is Pivotal for Cardiac Dyad Assembly and Intracellular Ca 2+ Dynamics. Circulation Research 2021, 128, 92-114, 10.1161/circresaha.119.315715.
- Andrew W. Senior; Richard Evans; John Jumper; James Kirkpatrick; Laurent Sifre; Tim Green; Chongli Qin; Augustin Žídek; Alexander W. R. Nelson; Alex Bridgland; et al.Hugo PenedonesStig PetersenKaren SimonyanSteve CrossanPushmeet KohliDavid T. JonesDavid SilverKoray KavukcuogluDemis Hassabis Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706-710, 10.1038/s41586-019-1923-7.
- Hong Su; Wenkai Wang; Zongyang Du; Zhenling Peng; Shang‐Hua Gao; Ming‐Ming Cheng; Jianyi Yang; Improved Protein Structure Prediction Using a New Multi‐Scale Network and Homologous Templates. Advanced Science 2021, 8, 2102592, 10.1002/advs.202102592.
- Jianchao Li; Haiyang Liu; Manmeet H. Raval; Jun Wan; Christopher M. Yengo; Wei Liu; Mingjie Zhang; Structure of the MORN4/Myo3a Tail Complex Reveals MORN Repeats as Protein Binding Modules. Structure 2019, 27, 1366-1374.e3, 10.1016/j.str.2019.06.004.

