Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | FEDERICA LAUDISI | + 5161 word(s) | 5161 | 2022-02-08 03:33:51 | | | |
| 2 | Vivi Li | Meta information modification | 5161 | 2022-02-17 10:56:18 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Laudisi, F. Intestinal Barrier Dysfunction in Gut Dysbiosis and Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/19574 (accessed on 23 July 2026).
Laudisi F. Intestinal Barrier Dysfunction in Gut Dysbiosis and Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/19574. Accessed July 23, 2026.
Laudisi, Federica. "Intestinal Barrier Dysfunction in Gut Dysbiosis and Diseases" Encyclopedia, https://encyclopedia.pub/entry/19574 (accessed July 23, 2026).
Laudisi, F. (2022, February 17). Intestinal Barrier Dysfunction in Gut Dysbiosis and Diseases. In Encyclopedia. https://encyclopedia.pub/entry/19574
Laudisi, Federica. "Intestinal Barrier Dysfunction in Gut Dysbiosis and Diseases." Encyclopedia. Web. 17 February, 2022.
Copy Citation
The intestinal mucosal barrier, also referred to as intestinal barrier, is widely recognized as a critical player in gut homeostasis maintenance as it ensures the complex crosstalk between gut microbes (both commensals and pathogens) and the host immune system. Highly specialized epithelial cells constantly cope with several protective and harmful agents to maintain the multiple physiological functions of the barrier as well as its integrity. However, both genetic defects and environmental factors can break such equilibrium, thus promoting gut dysbiosis, dysregulated immune-inflammatory responses, and even the development of chronic pathological conditions.
intestinal epithelial cells
mucosal barrier
microbiota
inflammatory bowel diseases
diet
mucus layer
aryl hydrocarbon receptor
cell commitment
junctional complexes
Paneth cells
1. Introduction
The intestinal mucosal barrier, also referred to as intestinal barrier, is a selectively permeable structure that grants the absorption of water, electrolytes, and essential dietary nutrients from the intestinal lumen into the circulation [1][2]. Apart from this role, the intestinal barrier mediates the crosstalk between commensal gut microbes and the host immunity and constitutes a first line of defence against intraluminal pathogenic antigens and potentially harmful microorganisms [1][2]. The intestinal barrier is composed of several elements that aid in its function as a physical and immunological defence boundary. These mainly include: (i) the outer mucus layer, encompassing the commensal gut microbiota, antimicrobial proteins (AMPs), and secretory immunoglobulin A (SIgA) molecules; (ii) the central single layer of specialized epithelial cells, derived from a pool of pluripotent stem cells at the base of the crypts that can be ultimately committed to goblet cells (which secrete mucins), Paneth cells (which synthesize antimicrobial peptides such as lysozyme and defensins), enteroendocrine cells (producing enteric hormones), enterocytes (absorbing water and nutrients), and Microfold cells, also referred to as M cells, (which are specialized for antigen sampling), following the up- or down-regulation of specific transcription factors; (iii) the inner lamina propria where cells from both innate (e.g., natural killer, neutrophils) and adaptive (e.g., T cells, B cells) immunity reside (Figure 1).
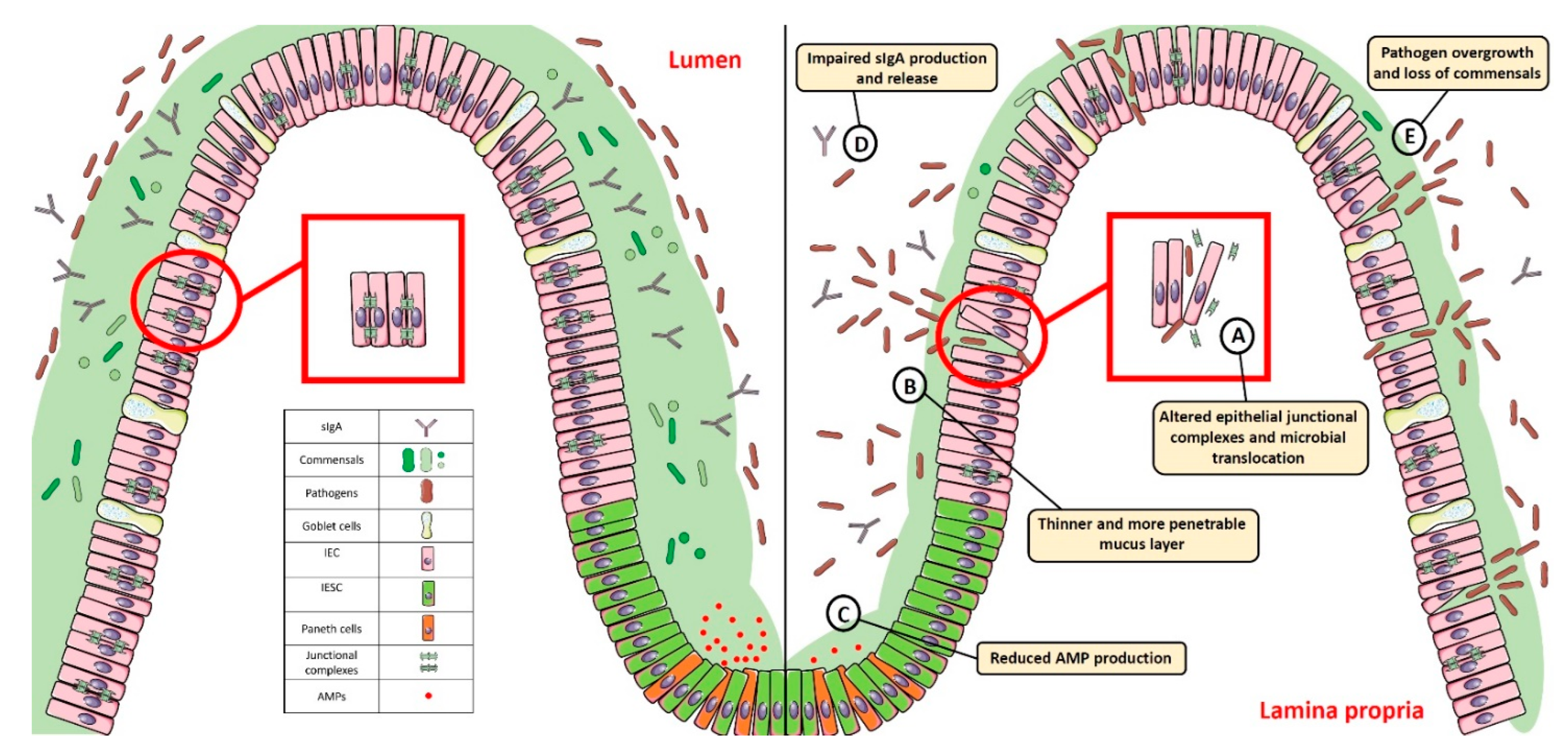
Figure 1. Gut homeostasis is established and maintained by the intestinal mucosal barrier. Alterations in its integrity and function, characterized by: (A) dysregulated junctional complexes, (B) thinner mucus layer, (C,D) reduced AMP and IgA production, and (E) pathogen overgrowth and penetration across the epithelial barrier, may perturb this fine balance and lead to gut dysbiosis. SIgA: Secretory Immunoglobulin A; AMP: antimicrobial peptide; IEC: intestinal epithelial cell; IESC: intestinal epithelial stem cell.
The ability to regulate the physiological processes occurring in the gut to keep internal states steady and balanced, also referred to as intestinal homeostasis, depends on complex interactions between the microbiota, the intestinal epithelium, and the host immune system. In particular, intestinal epithelial cells (IECs) act as frontline sensors for microbial encounters, and their hyporesponsiveness is ensured by the host innate immune system that can discriminate between signals derived from either commensal bacteria or pathogens [3][4][5]. Increasing evidence in germ-free mice highlighted the importance of commensal microbiome in maintaining gut homeostasis by providing protective, structural, and metabolic effects on the host mucosal surfaces [6][7]. For instance, commensals can release anti-microbial peptides, synthetize vitamins, contribute to ion adsorption and fermentation of non-digestible dietary residues, control epithelial cell differentiation, induce IgA secretion, and favour the immune system development [8][9]. Maintenance of such intestinal homeostasis also requires the structural integrity of the intestinal epithelium, which is ensured by junctional protein complexes (i.e., tight junctions, adherens junctions and desmosomes) that finely regulate intestinal permeability and seal adjacent epithelial cells [10] (Figure 1).
Dysfunction of the barrier physical integrity and/or an impaired function of the highly specialized cells composing the epithelial layer may lead to pathogen invasion and mucosal dysbiosis, resulting in a disruption of gut homeostasis that may ultimately trigger pathologic conditions, such as inflammatory bowel diseases (IBD), celiac disease, Clostridioides difficile infection (CDI), irritable bowel syndrome, colorectal cancer, type 1 diabetes, and obesity (Table 1) [11][12][13][14].
Table 1. Intestinal barrier alterations and related pathological conditions.
| Disease | Observation | Ref. |
|---|---|---|
| IBD |
|
[15][16][17][18][19] |
| Celiac disease |
|
[20][21][22][23][24] |
| CDI |
|
[25][26][27] |
| IBS |
|
[28][29][30][31][32][33][34] |
| CRC |
|
[35][36][37][38][39][40] |
| Obesity |
|
[41][42][43][44] |
| Type 1 diabetes |
|
[44][45][46][47][48][49][50] |
Abbreviations: IBD: Inflammatory Bowel Diseases; MyD88: Myeloid differentiation primary response 88; CXCR3: C-X-C Motif Chemokine Receptor 3; CDI: Clostridioides difficile infection; IBS: Irritable Bowel Syndrome; CRC: Colorectal cancer.
2. Breaking the Balance: Intestinal Barrier Dysfunction and Gut Dysbiosis
Both genetic defects and specific environmental factors are known to contribute to break the intestinal barrier balance and promote gut dysbiosis. In particular, impaired expression of genes related to cell commitment, junctional complexes, mucus production and secretion, Paneth cell activity, pathogen sensing, reactive oxygen species (ROS) production, xenobiotic response, and IgA secretion dramatically compromise intestinal epithelial barrier integrity and protective function (Table 2). Similarly, environmental factors—including bacterial infections; medication exposure (e.g., antibiotics) subsequent to pathogen infections or other diseases; and increased intake of high-fat compounds, sugars, and ethanol at the expense of fruits and vegetables—were reported to affect host microbiota composition and metabolic activities, leading to loss of commensals and overgrowth of pathogens (Table 3).
Table 2. Genetic defects affecting intestinal barrier homeostasis.
| Category | Gene | Effects on Intestinal Barrier | Ref. |
|---|---|---|---|
| Cell commitment | Hes1 | Reduced production of AMPs and mucus, gut dysbiosis, and inflammation. Precocious differentiation of Paneth cells. Impaired specification of IECs into enterocytes. | [51][52][53] |
| Math1 | Decreased frequency of goblet cells. | [54] | |
| Stk11 | Impaired released of AMPs and IL-18. Colitogenic bacteria overgrowth. | [55] | |
| Cdx2 | Altered mucus production and increased intestinal permeability and susceptibility to DSS-induced colitis. | [15][56] | |
| Gata6 | Impaired stem cell proliferation, reduction in villus length, Paneth cell, and enterocyte and enteroendocrine cell frequency. Increased number of goblet-like cells. Decreased levels of ZO-1 and increased intestinal permeability and susceptibility to experimental colitis and ileitis. Gut dysbiosis. | [57][58][59] | |
| Sox9 | Lack of differentiated Paneth cells, crypt enlargement, gut dysbiosis. | [60][61] | |
| Junctional Complexes | Jam-A/F11R | Increased intestinal permeability, low-grade intestinal inflammation, and increased susceptibility to DSS-induced colitis. | [62] |
| Cldn-2 and Cldn-15 double-KO | Impaired paracellular Na+ flow and malnutrition. | [63] | |
| Tjp1 | Apical surface brush border membrane and crevasses at intercellular junctions between enterocytes. Increased susceptibility to experimental colitis, delayed cell division, and mucosal healing. | [64][65] | |
| Ptpn2 | Increased claudin-2 expression, intestinal permeability, and inflammatory cytokine production. | [66] | |
| Mucus layer | Muc2 | ER stress and decreased frequency of goblet cells, altered mucus production, increased intestinal permeability, gut dysbiosis, and chronic intestinal inflammation. | [16][67][68] |
| Gfi1 | Accumulation of secretory progenitors, decrease in mucus and AMPs release. | [69] | |
| Foxo1 | Impaired mucus layer formation, overgrowth of mucin-degrading bacteria, and decrease of short-chain fatty acid-producing microbial species. Enhanced susceptibility to infection and inflammation. | [70] | |
| Paneth cells | Nod2 | Impaired α-defensins secretion. | [71][72] |
| Atg16l1 | Impaired autophagy in response to viral infection and decreased AMPs release. | [73][74] | |
| Xbp1 | Chronic ER stress in response to viral infection and decreased AMP release. | [75] | |
| Tcf4 | Reduced α-defensins secretion and CD development. | [76][77] | |
| PRRs | MyD88 | Increased stem cell proliferation. | [78] |
| Nod2 | Impaired pathogen sensing and clearance, and gut dysbiosis. No protection against oxidative stress-mediated cell death, and impaired epithelial regeneration. | [79][80][81][82][83] | |
| Oxidative Burst | Duox2 | Enhanced pathogen translocation to host lymphatic tissues. | [84] |
| Cyba | Decreased DUOX2 activity in response to Citrobacter rodentium. | [85] | |
| Xenobiotic Receptors | Pxr-Nr1I2 | Dysregulated TLR4-NF-κB signalling pathway, reduced ZO-1 and E-cadherin expression, increase in Claudin-2 levels. Higher susceptibility to IBD development. | [86][87][88] |
| AhR | Increased susceptibility to intestinal infection (Citrobacter rodentium), reduced mucus production, impaired tight junctions, and crypt hyperplasia. | [89][90][91] | |
| Secretory IgA | IgA | Gut dysbiosis. | [92] |
| pIgR | Impaired SIgA transcytosis across epithelial cells and gut dysbiosis. | [93][94] |
Abbreviations: Hes1: Hairy and enhancer of split 1; Math1: Mouse atonal homolog 1; Stk11: Serine threonine kinase 11; IL-18: Interleukin-18; Cdx2: Caudal type homeobox 2; DSS: Dextran Sodium Sulfate; Gata6: GATA binding factor 6; ZO1: Zonula Occludens-1; Sox9: SRY-Box Transcription Factor 9; Jam-A/F11R: Junctional adhesion molecules/F11 receptor; Cldn: claudin; Tjp1: Tight junction protein-1; Ptpn2: Protein tyrosine phosphatase non-receptor type 2; Muc2 Mucin-2; ER: Endoplasmic Reticulum; Gfi1: Growth factor independent 1; Foxo1: Forkhead box protein O1; Nod2: Nucleotide Binding Oligomerization Domain Containing 2; Atg16l1: Autophagy Related 16 Like 1; Xbp1: X-Box Binding Protein 1; Tcf4: transcription factor 4; CD: Crohn’s disease; MyD88: Myeloid differentiation primary response 88; Duox2: Dual oxidase 2; Cyba: Cytochrome B-245 Alpha Chain; Pxr/Nr1I2: Pregnane X receptor/Nuclear receptor subfamily 1 group I member 2; TLR4: Toll-like receptor-4; NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells; AhR: Aryl Hydrocarbon Receptor; IgA: Immunoglobulin A; pIgR: poly immunoglobulin receptor; AMP: Antimicrobial peptides; ZO-1: Zonula Occludens-1; ER: Endoplasmic reticulum; SIgA: Secretory immunoglobulin A.
Table 3. Environmental factors affecting intestinal barrier homeostasis.
| Category | Diet | Effects on Intestinal Barrier | Ref. |
| Junctional Complexes | Gluten | Alterations in adherent junctions and desmosomes and increased intestinal permeability and susceptibility to experimental colitis. Disassembly of ZO-1 from the tight junctional complex. | [20][95] |
| Glucose/Fructose | TJ and AJ proteins dysfunction, increased susceptibility to pathogen infection, gut dysbiosis, metabolic syndrome, oxidative stress, and chronic inflammation | [44][96] | |
| High-fat diet | ER stress in IECs, impairment of Claudin-1 expression and mucus barrier, and increased endotoxin serum levels. Increased taurocholic bile acid production and gut dysbiosis. | [97][98] | |
| Ethanol | Altered ZO-1 and occludin localization and impaired paracellular permeability. | [99][100] | |
| Mucus Layer | Fiber-deprived diet | Thinner mucus layer, gut dysbiosis, and chronic intestinal inflammation. | [67][101] |
| Paneth cells | Vitamin D deficiency and exposure to high-fat diet | Impaired expression of α-defensins, MMP7, and tight junction-related proteins; increased intestinal permeability; gut dysbiosis; and metabolic syndrome. Indiction of ER stress and secretion of misfolded α-defensins. | [102][103] |
| High fat diet | Decreased AMP expression, ER stress and autophagy induction, and gut dysbiosis. | [104] | |
| Western diet (deoxycholic acid) | Excessive activation of the farnesoid X receptor and type I interferon signalling pathways. | [105] | |
| Xenobiotic receptors | AhR ligand-free diet | Higher susceptibility to experimental colitis and gut dysbiosis. | [106][107] |
| Category | Bacterial infection | Effects on intestinal barrier | Ref. |
| Junctional Complexes | Infection by Salmonella typhimurium | Increased claudin-2 expression and bacterial invasion. | [108] |
| Infection by Vibrio cholerae | Zonula occludes toxin production and altered paracellular permeability. | [109][110] | |
| PRRs | LPS | Increased intestinal permeability. | [111] |
| Category | Medication exposure | Effects on intestinal barrier | Ref. |
| Junctional Complexes | Antibiotic treatment | Decreased production of microbial-derived short-chain fatty acids. Gut dysbiosis. Reduced ZO-1, occludin, and claudin-1 expression, and increased intestinal permeability. Altered microvilli morphology and reduced rate of intestinal epithelial cell turnover. | [112][113] |
Abbreviations: ZO-1: Zonula Occludens-1; TJ: Tight junction; AJ: Adherent junction; ER: Endoplasmic Reticulum; IECs: Intestinal Epithelial Cells; MMP: Matrix Metallopeptidase; AMP: Antimicrobial peptide; AhR: aryl hydrocarbon receptor; PRR: Pattern recognition receptors; LPS: lipopolysaccharide; IECs: intestinal epithelial cells.
2.1. Impairment of Cell Commitment
As previously anticipated, the intestinal barrier is characterized by a self-renewing epithelium, organized in crypts and villi, including both stem cells and differentiated cells. As the epithelial barrier has to deal with multiple physiologic activities, it requires different specialized cells, some of which are able to produce and secrete several molecules—such as antimicrobial peptides, mucins and hormones—and others that are able to adsorb water and nutrients. To achieve this goal, stem cells, after multiple transit-amplifying (TA) divisions, terminally differentiate into either secretory or absorptive lineages depending on the tightly regulated expression/inhibition of specific transcription factors [114][115]. For instance, the cell surface receptor Notch drives the cell commitment process by binding to the Notch ligands Deltalike (Dll) and Jagged families [116]. A cell expressing the Notch ligands will differentiate into a secretory cell (e.g., goblet cell, Paneth cell) upon expression of the Atonal BHLH Transcription Factor 1 (Atoh1), whereas a neighbour cell expressing the activated Notch receptor will induce the expression of the target gene hairy and enhancer of split 1 (Hes1)—a Atoh1 inhibitor—and will differentiate into an absorptive cell (e.g., enterocyte) [54]. Obviously, a dysregulated expression of the above-mentioned proteins definitely compromises the intestinal epithelial cell commitment and, in turn, the intestinal barrier integrity and function, as demonstrated by the association of several polymorphisms in genes encoding commitment-related transcription factors with intestinal barrier dysfunctions and intestinal inflammatory diseases (Figure 2) [51][52][53][54][55]. In this context, by employing genetically engineered mouse models, Guo and colleagues demonstrated that deficiency of the Hes1 gene in IECs negatively affected antimicrobial peptides and mucus production, thus resulting in gut dysbiosis and inflammation [51]. Along the same line was the demonstration that mice deficient for Math1 (also referred to as Atoh1) displayed complete abrogation of goblet cells in the intestine [54]. Similar results were observed in mice with conditional deletion of the serine threonine kinase 11 (Stk11) gene, involved in the differentiation of stem cells into the secretory lineage cell types (e.g., goblet cells and Paneth cells), in IECs [55]. These animals displayed increased susceptibility to gut inflammation in association with reduced production of antimicrobial peptides and IL-18, as well as an uncontrolled expansion of colitogenic bacteria [55].
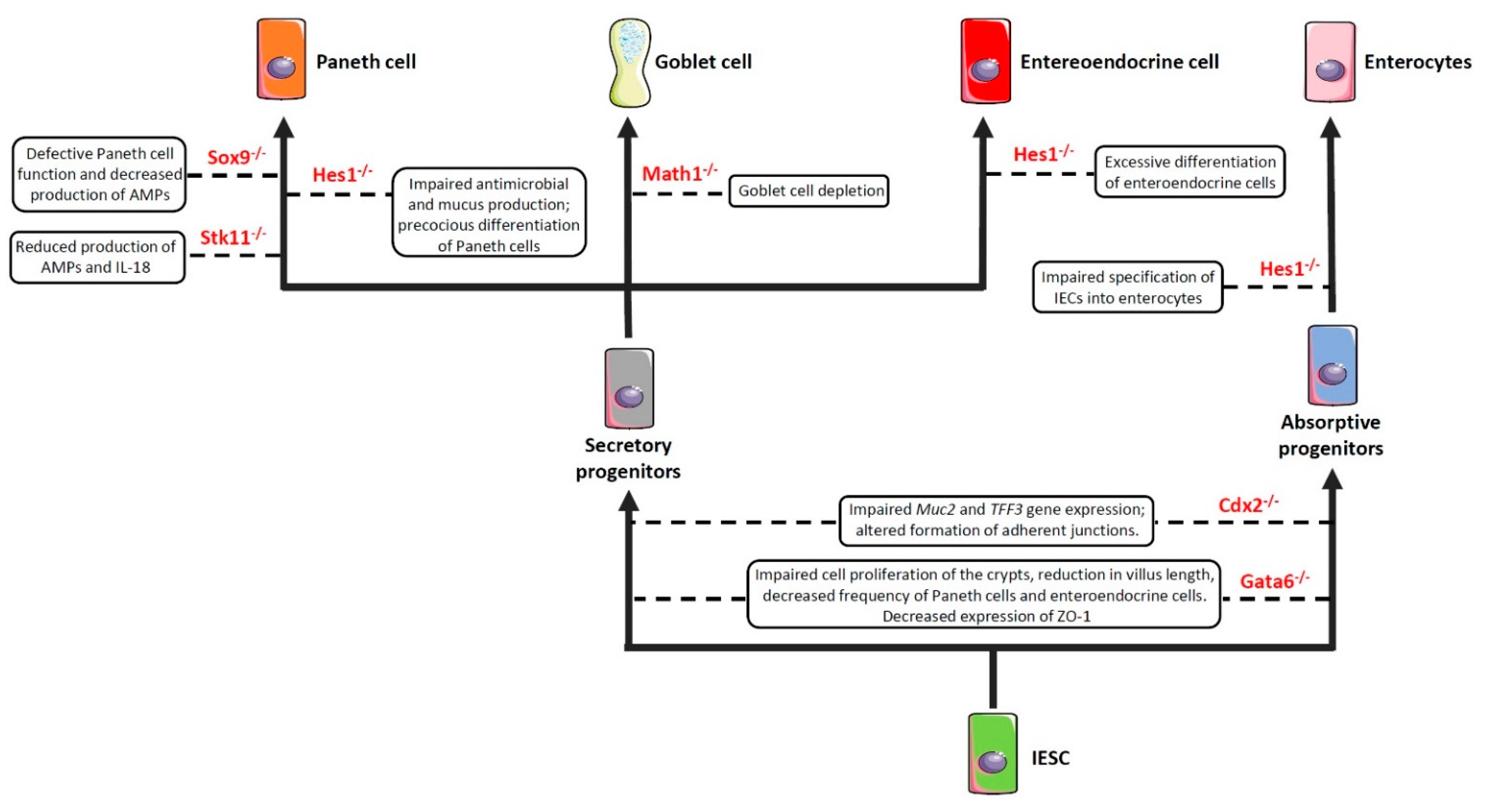
Figure 2. Impaired expression of genes encoding commitment-related transcription factors compromises epithelial cell differentiation and intestinal barrier function. Boxes enclose the effect/s of the knockdown of the genes depicted in red on the indicated cell commitment. Abbreviations: Sox9: SRY-Box Transcription Factor 9; Hes1: Hairy and enhancer of split 1; Stk11: Serine threonine kinase 11; Math1: Mouse atonal homolog 1; Cdx2: Caudal type homeobox 2; Gata6: GATA binding factor 6; Muc2: Mucin 2; TFF3: Trefoil factor 3; ZO-1: Zonula Occludens-1; AMPs: Antimicrobial peptides; IESC: Intestinal epithelial stem cells.
Another important transcription factor involved in the commitment of IECs is the caudal type homeobox 2 (Cdx2) gene [117]. Indeed, CDX2 is a positive regulator of the Muc2 and Trefoil Factor 3 (TFF3) genes [56][118] involved in the production and stabilization of the mucus layer, respectively, and whose deficiency induces hypersensitivity to chemically-induced colitis (such as that induced by dextran sulfate sodium (DSS)) [15][56]. In addition, CDX2 controls cell-cell interactions and the expression of cadherins, which are important in the formation of the adherens junctions [119][120][121]. In support of this view is the evidence that mice bearing one non-functional Cdx2 allele (Cdx2+/− mice) displayed increased intestinal permeability and were more susceptible to the abrasive effect of DSS [117].
The GATA binding factor 6 (GATA6) is a zinc finger transcription factor that regulates cell proliferation, differentiation, and gene expression in several tissues [122]. For instance, GATA6 is involved in cell proliferation and differentiation along the gastrointestinal tract [57][58]. In particular, conditional deletion of Gata6 in IECs resulted in impaired cell proliferation of the crypts, reduction in villus length, decreased frequency of Paneth cells and enteroendocrine cells, increased number of goblet-like cells, and dysregulated expression of enterocyte-related genes in the ileum [57]. Similar alterations were observed in the colon, where Gata6 deficiency affected stem cell proliferation and differentiation into Paneth cells, enteroendocrine cells, and enterocytes [58]. Researchers' study has recently demonstrated that conditional deletion of Gata6 in the gut epithelium significantly affected intestinal barrier integrity, leading to decreased expression of the tight junction-related protein zonula occludens-1 (ZO-1), and resulting in increased paracellular permeability, microbial dysbiosis, and susceptibility to gut inflammation [59]. Interestingly, researchers also reported a decreased expression of GATA6 in the intestinal epithelium of IBD patients, thus suggesting that a reduced expression of this transcription factor may contribute to intestinal barrier dysfunction in these subjects [59]. In the intestinal epithelium, defects in Paneth cell function—and the consequent decrease in the antimicrobial peptide production—may also result from the deletion of Sox9 [60]. Indeed, by generating mice that harbored a conditional Sox9 gene and a Villin-Cre transgene, Mori-Akiyama et al. reported that lack of Sox9 expression in the intestinal epithelium of Sox9fl/fl Villin-Cre+ mice resulted in the complete absence of differentiated Paneth cells, although the differentiation of other intestinal epithelial cell subsets (e.g., goblet cells, enterocytes) was not affected. Moreover, Sox9 deficiency also lead to crypt enlargement, a marked increase in cell proliferation throughout the crypts, as well as a replacement of the Paneth cells by proliferating epithelial cells [60]. More recently, by employing the same conditional mouse model, Riba and colleagues showed that Sox9 deletion in the intestinal epithelium reduced lysozyme production. This effect resulted in significant microbial dysbiosis, characterized by E. coli overgrowth and ultimately leading to visceral hypersensitivity [61].
2.2. Impairment of Epithelial Junctional Complexes
The intestinal epithelial barrier’s main function is to protect the host from luminal antigens, pathogens, and toxins, while allowing selective permeability to water, nutrients, and electrolytes. In particular, transcellular permeability, involved in solute transport through the epithelial cells, is mediated by selective transporters for amino acids, electrolytes, short chain fatty acids, and sugars [123]. Paracellular permeability, instead, occurs through intercellular junctional complexes encompassing adherens junctions (AJs), tight junctions (TJs), and desmosomes [124][125]. These transmembrane proteins, localized both at the apical-lateral membrane junction and along the lateral membrane, mediate the contact between adjacent IECs, thus sealing the intracellular spaces. The AJs (e.g., catenins, cadherins) and desmosomes (e.g., desmoglein, desmocollins) regulate the mechanical linkage of adjacent cells, while the TJs (e.g., ZO-1, claudin-2, occludins, Junctional Adhesion Molecule) form an apical junctional complex that seals the intercellular space and modulates selective paracellular permeability [124][125][126][127][128].
Alterations in the formation/distribution of the intercellular junctional complexes, which may occur in men with specific genetic susceptibilities, as well as in response to dietary factors and bacterial infections, may result in intestinal epithelial barrier breakdown and translocation of the luminal content into the lamina propria, leading to gut dysbiosis, uncontrolled immune/inflammatory responses, and, ultimately, pathological conditions [10].
For instance, gliadin (a glycoprotein representing the major component of wheat gluten) has been reported to deeply affect the expression and distribution of several junctional complexes in the small intestine of celiac patients by binding to CXC motif receptor 3 (CXCR3) on epithelial cells [20]. This interaction induces the release of zonulin, a human protein analogue of the Zonula occludens toxin (ZOT) from Vibrio cholerae, through the recruitment of Myeloid differentiation primary response (MyD)-88 [20][21][129][130]. Increased levels of zonulin were detected in the intestinal tissues taken from celiac disease patients during the acute phase compared to those taken from healthy controls. Once released, Zonulin leads to transactivation of EGF receptor (EGFR) via proteinase-activated receptor 2 (PAR2) activation in the intestinal epithelium and subsequent tight junction disassembly [20][21][129][130].
The impairment of the epithelial junctional complexes importantly contributes to the development of other chronic inflammatory conditions, such as IBD. For example, increased expression of claudin-2, as well as impaired expression and redistribution of claudin-5, -8 and occludin were reported in Crohn’s disease patients, leading to increased intestinal permeability and bacterial translocation [131]. A similar severe condition was described also in the colonic mucosa of patients with ulcerative colitis, in association with the dysregulated expression of occludin, ZO-1, claudin-1, JAM, beta-catenin, E-cadherin, and the consequent transepithelial migration of neutrophils [132].
The above-mentioned chronic inflammatory conditions importantly contribute to the development and progress of colorectal carcinogenesis. Interestingly, increased expression of claudin-1 and claudin-2 was found to correlate with inflammatory activity, IBD-associated dysplasia, and sporadic adenomas [36]. Similarly, Dhawan et al. observed that claudin-1 expression was increased in colon carcinomas and metastatic lesions and played a key role for tumorigenesis and invasiveness of colonic epithelial cells [35]. Claudin-2 was also reported to be increased in tissues taken from CRC and IBD-associated CRC patients and to promote and sustain cell proliferation and tumor growth in cultured cells and experimental models [133].
Dysfunctions of the epithelial junctional complexes and the consequent increase of intestinal permeability and gut dysbiosis correlate with the development and progression of other pathological conditions. In particular, increased intestinal permeability was seen to precede and/or to be an early biomarker of diabetes development in patients, as well as in experimental models of the disease [45][134][135]. Moreover, increased serum levels of zonulin, in association with altered intestinal permeability, were described in a subgroup of patients with type 1 diabetes and their first-degree relatives, suggesting this molecule as a valid early biomarker of disease development [49].
Animal models employing genetically engineered mice have helped to better understand the link between junctional complex dysregulation and the development of dysbiosis and pathologic conditions. In this context, Laukoetter et al. reported a role for Junctional Adhesion Molecule (JAM)-A, a TJ component contributing to the control of barrier function and leukocyte migration, in regulating intestinal permeability and inflammation in vivo [62]. Indeed, despite showing normal epithelial architecture, JAM-A knockout mice developed low-grade colonic inflammation (characterized by enhanced polymorphonuclear leukocyte infiltration and large lymphoid aggregates not seen in sham mice) [62]. Barrier function experiments revealed increased mucosal permeability, as indicated by enhanced dextran flux, and decreased transepithelial electrical resistance in JAM-A knockout mice compared to wild-type control mice [62]. Consistently, JAM-A deficiency increased the permeability of in vitro monolayers derived from the human colonic epithelial cell line SK-CO15 compared with control. Moreover, JAM-A deficient mice were more susceptible to the DSS-driven experimental colitis compared to controls, although the colonic mucosa showed less injury and increased epithelial proliferation [62]. Analyses of other TJ-related proteins showed increased expression of claudin-10 and -15, both of which tune TJ barrier function by the formation of ion-selective pores, following JAM-A knockdown in the colonic mucosa of mice and in SK-CO15 cell monolayers [62].
In a later article, Wada and colleagues reported that mice with the double knockdown of claudin-2 (Cldn-2) and claudin-15 (Cldn-15) genes had impaired paracellular Na+ flow and subsequent malnutrition, leading to infant death [63].
By employing cultured epithelial cells and an intestinal epithelial-specific knockout mouse (that is, Tjp1fl/fl Villin-Cre+ mouse), Odenwald and co-workers showed that the TJ scaffolding protein ZO-1 was essential for development of unified apical surfaces in vitro and in vivo. In detail, conditional deletion of ZO-1 in IECs of Tjp1fl/fl Villin-Cre+ mice did not significantly alter crypt-villus architecture, whereas it affected apical tissue continuity, which is by characterized apical surface brush border membrane, and the presence of crevasses at intercellular junctions between enterocytes, likely by modulating actomyosin contraction and membrane traffic [64]. Recently, Kuo and colleagues reported decreased ZO-1 expression, both at RNA and protein level, in intestinal mucosal biopsies isolated from IBD patients as compared with those isolated from healthy controls [65]. Loss of ZO-1 expression in epithelial cells in Tjp1fl/fl Villin-Cre+ mice did not promote spontaneous disease, but it exacerbated tissue damage and weight loss during experimental colitis, as well as delayed the mucosal healing [65]. The authors also reported that ZO-1 is critically involved in the cell division phase upon damage. In particular, by associating with the centriole and mitotic spindle, ZO-1 contributed to both Wnt–β-catenin signaling and mitotic spindle orientation, suggesting that ZO-1 may actively contribute to the intestinal epithelial barrier restoration [65]. In line with these observations, researchers recently found that loss of Gata6 expression in IECs of genetically engineered mice resulted in increased intestinal permeability, gut dysbiosis, and microbial-driven intestinal inflammation. These effects were associated with decreased ZO-1 expression and epithelial damage both in the ileum and colon. Experiments in cultured cells suggested that ZO-1 expression could be directly modulated by GATA6 [59]. Recently, Marchelletta and colleagues reported that the impaired function of T cell protein tyrosine phosphatase (TCPTP), encoded by the protein tyrosine phosphatase non-receptor type 2 (PTPN2) gene, contributed to the epithelial tight junction protein remodeling and increased intestinal permeability [66]. In particular, Tcptp-deficient mice showed increased claudin-2 expression, intestinal permeability, and inflammatory cytokine production [66]. In detail, TCPTP was able to maintain the localization of ZO-1 and occludin at apical tight junctions, as well as to modulate the turn-over of claudin-2, a cation pore-forming transmembrane protein, by upregulating the serine metalloproteinase matriptase, which promoted claudin-2 proteosomal degradation [66].
Apart from defects in the above-mentioned epithelial junction-related molecules, several dietary factors may contribute to increase intestinal permeability and trigger/amplify pathologic conditions [136]. A good example in this regard is given by gluten, which, in addition to its well-known detrimental effects on barrier integrity and TJ protein activity in celiac disease, can actively promote dysregulation of intestinal barrier function in non-celiac patients. Of note, mice exposed to a gluten-rich diet showed alterations in adherent junctions and desmosomes, resulting in increased intestinal permeability and susceptibility to DSS-driven experimental colitis [95].
Glucose and fructose are additional macronutrients found to trigger TJ and AJ protein dysfunction, thus promoting changes in microbiota composition, increased susceptibility to pathogen infection, as well as metabolic syndrome [44]. In mouse experimental models, uncontrolled metabolism of fructose in the liver and in the small intestine, due to the excessive delivery of this sugar (15% in water for 3 weeks), induced the transcriptional expression of fructokinase (a protein involved in fructose metabolism), TJ alterations, energy depletion, oxidative stress, and chronic inflammation [96]. On the other hand, mice deficient of the fructokinase isoforms A and C (KHK-A, KHK-C) were protected from such detrimental effects. Notably, loss of KHK-A function only did not prevent alterations in TJs, thus suggesting that intestinal barrier impairment was mainly mediated by KHK-C activity [96].
Detrimental effects of dietary fats on the epithelial junctional complexes have been also reported by several studies. In particular, mice exposed to a high-fat diet for 3, 11, and 22 weeks showed induction of endoplasmic reticulum (ER) stress in IECs, as well as an impairment of Claudin-1 expression and mucus barrier, with the consequent increase of endotoxin serum levels and gut dysbiosis [97]. Similarly, Devkota and colleagues demonstrated that the increased availability of taurocholic bile acid, due to the consumption of a diet high in saturated (milk derived)-fat, promoted the expansion of the low abundance pathobiont Bilophila wadsworthia (a member of the Deltaproteobacteria), which, in turn, was able to impair intestinal barrier integrity in genetically susceptible Il-10−/− mice due to its sulphite-reducing activity [98]. Another dietary habit found to affect TJ activity is ethanol consumption. Exposure to non-cytotoxic doses of ethanol (as those detected in the blood of moderate drinkers) impaired paracellular permeability in vitro due to alterations in ZO-1 and occludin localization [99][100].
Both localization and activity of epithelial junctional complexes can also be affected by pathogen invasion and toxin secretion. For instance, Salmonella typhimurium was found to up-regulate the colonic expression of the leaky protein claudin-2, which plays an opposite role in the modulation of intestinal permeability compared to other TJ proteins involved in barrier maintenance, thus facilitating bacterial invasion [108]. Vibrio cholerae, instead, was reported to target the intestinal epithelial barrier by producing the zonula occludens toxin (ZOT), which transiently affects the paracellular permeability in the small intestine by opening TJs through a protein kinase C-dependent actin reorganization [109][110].
On its side, antibiotic treatment dramatically influences intestinal permeability by compromising host microbial ecology. In particular, mice exposed to antibiotics for 2 weeks developed mucosal dysbiosis characterized by decreased production of short-chain fatty acids, such as butyrate (known to sustain barrier function and integrity), by commensals [112]. Moreover, antibiotic treatment hampered intestinal TJ function and increased intestinal permeability by reducing the expression of ZO-1, occluding, and claudin-1 [112]. Similar results were obtained in antibiotic-treated germ-free mice, which presented altered microvilli morphology and reduced rate of intestinal epithelial cell turnover compared to sham mice [113]. Altogether, these results indicate a key role for commensal microbiota in preserving epithelial junctional complexes and gut barrier integrity, highlighting a possible detrimental effect of antibiotic exposure on such a fine balance.
2.3. Thinning/Depletion of the Mucus Layer
Goblet cells are specialized IECs able to synthetize and secrete mucin proteins into the lumen. Mucin proteins are pivotal in creating a protective mucus layer acting against pathogens, chemicals, and mechanical stress in order to maintain gut homeostasis and protect the inner mucosal surface [137]. The mucus layer, mainly composed of water, electrolytes, lipids, and glycosylated mucins [138], represents an important source of antimicrobial peptides and immunoglobulins and can directly interact with commensals, providing nutrients and attachment sites depending on the mucin glycosylation profile [139].
Mucolytic bacteria (e.g., Akkermansia muciniphila, Bacteroides thetaiotaomicron, Ruminococcus gnavus, Ruminococcus torques) represent an important class of commensals as they are able to digest glycans (from dietary fibers) and mucins through glycosidase enzymes, and to produce, in turn, short chain fatty acids (such as acetate and butyrate) acting as energy source for colonocytes and contributing to protect the intestinal barrier integrity [139]. However, the fine balance between goblet cell-mediated replenishment of mucus and its degradation by commensals can be affected by a fiber-deprived diet, as indicated by the fact that mice subjected to intermittent dietary fiber deprivation presented a thinner mucus layer due to O-linked glycan digestion by the fiber-deprived microbiota [101]. Thus, enrichment in mucus-degrading bacteria may impair the mucus layer thickness and viscosity and promote enteric pathogens adherence and penetration, ultimately causing gut dysbiosis and chronic intestinal inflammation [67][101]. These pathological alterations were observed in the Winnie murine model of spontaneous colitis, characterized by a missense mutation in the Muc2 gene [16][67]. The phenotype of Winnie mice was characterized by altered mucus production as early as 4 weeks of age, with ensuing intestinal barrier dysfunction, gut dysbiosis, and inflammation [16][67]. In particular, impaired Muc2 expression affected the number of goblet cells, which underwent unresolved ER stress and accumulation of mucin precursors [16][67]. All these processes were associated with apoptotic cell death, increased intestinal permeability, pathogen penetration into the inner mucus layer, and adherence to epithelial cells, as well as bacterial translocation into the lamina propria [16][67]. The subsequent uncontrolled immune response towards pathogens (e.g., enhanced dendritic cell activation, T-helper cytokine production) promoted chronic intestinal inflammation and gut dysbiosis, characterized by the outgrowth of Bacteroidetes and Verrucomicrobia (such as Akkermansia muciniphila) [67][68].
Impaired mucus layer integrity can also depend on mutations in the Gfi1 gene. Gfi1 functions downstream of Math1 in the intestinal epithelium and encodes molecules involved in the stem cell differentiation into the different secretory cell lineages [69]. In particular, Gfi1-deficient mice displayed alterations in terminal differentiation and morphology of goblet cells and Paneth cells, together with accumulation of immature secretory progenitors, as well as a decrease in mucin and antimicrobial peptide release [69]. Recently, the Foxo1 trascription factor was described to be critically involved in mucin granule release through autophagy [70]. In particular, Foxo1fl/fl Villin-Cre+ mice showed impaired mucus layer formation and subsequent dysbiosis, resulting in disrupted intestinal barrier integrity and enhanced susceptibility to infection and tissue inflammation. Moreover, Foxo1 deficiency in IECs resulted in the overgrowth of mucin-degrading bacteria and a decrease of short-chain fatty acid-producing microbial species, which further affected the intestinal barrier function [70].
In addition to mucus production and degradation, gut microbiota composition is able to influence the mucus properties. In this regard, Jakobsson and colleagues reported that the mucus layer of germ-free mice was characterized by a higher mucus penetrability as compared to conventional mice [111]. Moreover, mice with identical genetic background, but hosted in two rooms of the same specific pathogen-free animal facility, showed different mucus properties, evidenced by the fact that one colony had an impenetrable inner mucus layer, whereas the other showed opposite features [111]. The authors suggested that these differences relied on changes in the gut microbiota composition as the different mucus phenotypes were acquired by germ-free mice upon faecal microbiota transplantation [111]. In particular, mice with an impenetrable inner mucus layer showed increased frequency of the Erysipelotrichi class, whereas Proteobacteria and TM7 expanded in mice with more penetrable mucus [111]. Hence, even genetically identical animals housed in the same facility may have distinct microbiotas and barrier structures [111].
Taken together, these results highlight the mutualistic effects between the gut microbial community and the mucus layer and their consequences on intestinal barrier integrity and function.
References
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153.
- Goto, Y.; Kiyono, H. Epithelial barrier: An interface for the cross-communication between gut flora and immune system. Immunol. Rev. 2012, 245, 147–163.
- Okumura, R.; Takeda, K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp. Mol. Med. 2017, 49, e338.
- Hooper, L.V.; Macpherson, A.J. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol. 2010, 10, 159–169.
- Martens, E.C.; Neumann, M.; Desai, M.S. Interactions of commensal and pathogenic microorganisms with the intestinal mucosal barrier. Nat. Rev. Microbiol. 2018, 16, 457–470.
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Nunez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89.
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323.
- Hayes, C.L.; Dong, J.; Galipeau, H.J.; Jury, J.; McCarville, J.; Huang, X.; Wang, X.Y.; Naidoo, A.; Anbazhagan, A.N.; Libertucci, J.; et al. Commensal microbiota induces colonic barrier structure and functions that contribute to homeostasis. Sci. Rep. 2018, 8, 14184.
- Ghosh, S.; Whitley, C.S.; Haribabu, B.; Jala, V.R. Regulation of Intestinal Barrier Function by Microbial Metabolites. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 1463–1482.
- Su, L.; Shen, L.; Clayburgh, D.R.; Nalle, S.C.; Sullivan, E.A.; Meddings, J.B.; Abraham, C.; Turner, J.R. Targeted epithelial tight junction dysfunction causes immune activation and contributes to development of experimental colitis. Gastroenterology 2009, 136, 551–563.
- Groschwitz, K.R.; Hogan, S.P. Intestinal barrier function: Molecular regulation and disease pathogenesis. J. Allergy Clin. Immunol. 2009, 124, 3–20.
- Mankertz, J.; Schulzke, J.D. Altered permeability in inflammatory bowel disease: Pathophysiology and clinical implications. Curr. Opin. Gastroenterol. 2007, 23, 379–383.
- Martini, E.; Krug, S.M.; Siegmund, B.; Neurath, M.F.; Becker, C. Mend Your Fences: The Epithelial Barrier and its Relationship with Mucosal Immunity in Inflammatory Bowel Disease. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 33–46.
- Gitter, A.H.; Wullstein, F.; Fromm, M.; Schulzke, J.D. Epithelial barrier defects in ulcerative colitis: Characterization and quantification by electrophysiological imaging. Gastroenterology 2001, 121, 1320–1328.
- Van der Sluis, M.; De Koning, B.A.; De Bruijn, A.C.; Velcich, A.; Meijerink, J.P.; Van Goudoever, J.B.; Buller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006, 131, 117–129.
- Heazlewood, C.K.; Cook, M.C.; Eri, R.; Price, G.R.; Tauro, S.B.; Taupin, D.; Thornton, D.J.; Png, C.W.; Crockford, T.L.; Cornall, R.J.; et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008, 5, e54.
- Braun, A.; Treede, I.; Gotthardt, D.; Tietje, A.; Zahn, A.; Ruhwald, R.; Schoenfeld, U.; Welsch, T.; Kienle, P.; Erben, G.; et al. Alterations of phospholipid concentration and species composition of the intestinal mucus barrier in ulcerative colitis: A clue to pathogenesis. Inflamm. Bowel Dis. 2009, 15, 1705–1720.
- Larsson, J.M.; Karlsson, H.; Crespo, J.G.; Johansson, M.E.; Eklund, L.; Sjovall, H.; Hansson, G.C. Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm. Bowel Dis. 2011, 17, 2299–2307.
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Burgel, N.; Fromm, M.; et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005, 129, 550–564.
- Lammers, K.M.; Lu, R.; Brownley, J.; Lu, B.; Gerard, C.; Thomas, K.; Rallabhandi, P.; Shea-Donohue, T.; Tamiz, A.; Alkan, S.; et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology 2008, 135, 194–204.e3.
- Fasano, A.; Not, T.; Wang, W.; Uzzau, S.; Berti, I.; Tommasini, A.; Goldblum, S.E. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet 2000, 355, 1518–1519.
- El Asmar, R.; Panigrahi, P.; Bamford, P.; Berti, I.; Not, T.; Coppa, G.V.; Catassi, C.; Fasano, A. Host-dependent zonulin secretion causes the impairment of the small intestine barrier function after bacterial exposure. Gastroenterology 2002, 123, 1607–1615.
- Szakal, D.N.; Gyorffy, H.; Arato, A.; Cseh, A.; Molnar, K.; Papp, M.; Dezsofi, A.; Veres, G. Mucosal expression of claudins 2, 3 and 4 in proximal and distal part of duodenum in children with coeliac disease. Virchows Arch. Int. J. Pathol. 2010, 456, 245–250.
- Ciccocioppo, R.; Finamore, A.; Ara, C.; Di Sabatino, A.; Mengheri, E.; Corazza, G.R. Altered expression, localization, and phosphorylation of epithelial junctional proteins in celiac disease. Am. J. Clin. Pathol. 2006, 125, 502–511.
- Theriot, C.M.; Koenigsknecht, M.J.; Carlson, P.E., Jr.; Hatton, G.E.; Nelson, A.M.; Li, B.; Huffnagle, G.B.; Li, J.Z.; Young, V.B. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun. 2014, 5, 3114.
- Theriot, C.M.; Young, V.B. Microbial and metabolic interactions between the gastrointestinal tract and Clostridium difficile infection. Gut Microbes 2014, 5, 86–95.
- Kachrimanidou, M.; Tsintarakis, E. Insights into the Role of Human Gut Microbiota in Clostridioides difficile Infection. Microorganisms 2020, 8, 200.
- Hanning, N.; Edwinson, A.L.; Ceuleers, H.; Peters, S.A.; De Man, J.G.; Hassett, L.C.; De Winter, B.Y.; Grover, M. Intestinal barrier dysfunction in irritable bowel syndrome: A systematic review. Ther. Adv. Gastroenterol. 2021, 14, 1756284821993586.
- Martinez, C.; Vicario, M.; Ramos, L.; Lobo, B.; Mosquera, J.L.; Alonso, C.; Sanchez, A.; Guilarte, M.; Antolin, M.; de Torres, I.; et al. The jejunum of diarrhea-predominant irritable bowel syndrome shows molecular alterations in the tight junction signaling pathway that are associated with mucosal pathobiology and clinical manifestations. Am. J. Gastroenterol. 2012, 107, 736–746.
- Martinez, C.; Lobo, B.; Pigrau, M.; Ramos, L.; Gonzalez-Castro, A.M.; Alonso, C.; Guilarte, M.; Guila, M.; de Torres, I.; Azpiroz, F.; et al. Diarrhoea-predominant irritable bowel syndrome: An organic disorder with structural abnormalities in the jejunal epithelial barrier. Gut 2013, 62, 1160–1168.
- Martinez, C.; Rodino-Janeiro, B.K.; Lobo, B.; Stanifer, M.L.; Klaus, B.; Granzow, M.; Gonzalez-Castro, A.M.; Salvo-Romero, E.; Alonso-Cotoner, C.; Pigrau, M.; et al. miR-16 and miR-125b are involved in barrier function dysregulation through the modulation of claudin-2 and cingulin expression in the jejunum in IBS with diarrhoea. Gut 2017, 66, 1537–1538.
- Dunlop, S.P.; Hebden, J.; Campbell, E.; Naesdal, J.; Olbe, L.; Perkins, A.C.; Spiller, R.C. Abnormal intestinal permeability in subgroups of diarrhea-predominant irritable bowel syndromes. Am. J. Gastroenterol. 2006, 101, 1288–1294.
- Long, Y.; Du, L.; Kim, J.J.; Chen, B.; Zhu, Y.; Zhang, Y.; Yao, S.; He, H.; Zheng, X.; Huang, Z.; et al. MLCK-mediated intestinal permeability promotes immune activation and visceral hypersensitivity in PI-IBS mice. Neurogastroenterol. Motil. Off. J. Eur. Gastrointest. Motil. Soc. 2018, 30, e13348.
- Zhou, Q.; Verne, M.L.; Fields, J.Z.; Lefante, J.J.; Basra, S.; Salameh, H.; Verne, G.N. Randomised placebo-controlled trial of dietary glutamine supplements for postinfectious irritable bowel syndrome. Gut 2019, 68, 996–1002.
- Dhawan, P.; Singh, A.B.; Deane, N.G.; No, Y.; Shiou, S.R.; Schmidt, C.; Neff, J.; Washington, M.K.; Beauchamp, R.D. Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer. J. Clin. Investig. 2005, 115, 1765–1776.
- Weber, C.R.; Nalle, S.C.; Tretiakova, M.; Rubin, D.T.; Turner, J.R. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab. Investig. A J. Tech. Methods Pathol. 2008, 88, 1110–1120.
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215.
- Buc, E.; Dubois, D.; Sauvanet, P.; Raisch, J.; Delmas, J.; Darfeuille-Michaud, A.; Pezet, D.; Bonnet, R. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS ONE 2013, 8, e56964.
- Boleij, A.; Hechenbleikner, E.M.; Goodwin, A.C.; Badani, R.; Stein, E.M.; Lazarev, M.G.; Ellis, B.; Carroll, K.C.; Albesiano, E.; Wick, E.C.; et al. The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2015, 60, 208–215.
- Biarc, J.; Nguyen, I.S.; Pini, A.; Gosse, F.; Richert, S.; Thierse, D.; Van Dorsselaer, A.; Leize-Wagner, E.; Raul, F.; Klein, J.P.; et al. Carcinogenic properties of proteins with pro-inflammatory activity from Streptococcus infantarius (formerly S.bovis). Carcinogenesis 2004, 25, 1477–1484.
- Genser, L.; Aguanno, D.; Soula, H.A.; Dong, L.; Trystram, L.; Assmann, K.; Salem, J.E.; Vaillant, J.C.; Oppert, J.M.; Laugerette, F.; et al. Increased jejunal permeability in human obesity is revealed by a lipid challenge and is linked to inflammation and type 2 diabetes. J. Pathol. 2018, 246, 217–230.
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481.
- Pendyala, S.; Walker, J.M.; Holt, P.R. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology 2012, 142, 1100–1101.e2.
- Thaiss, C.A.; Levy, M.; Grosheva, I.; Zheng, D.; Soffer, E.; Blacher, E.; Braverman, S.; Tengeler, A.C.; Barak, O.; Elazar, M.; et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 2018, 359, 1376–1383.
- Meddings, J.B.; Jarand, J.; Urbanski, S.J.; Hardin, J.; Gall, D.G. Increased gastrointestinal permeability is an early lesion in the spontaneously diabetic BB rat. Am. J. Physiol. 1999, 276, G951–G957.
- Neu, J.; Reverte, C.M.; Mackey, A.D.; Liboni, K.; Tuhacek-Tenace, L.M.; Hatch, M.; Li, N.; Caicedo, R.A.; Schatz, D.A.; Atkinson, M. Changes in intestinal morphology and permeability in the biobreeding rat before the onset of type 1 diabetes. J. Pediatric Gastroenterol. Nutr. 2005, 40, 589–595.
- Bosi, E.; Molteni, L.; Radaelli, M.G.; Folini, L.; Fermo, I.; Bazzigaluppi, E.; Piemonti, L.; Pastore, M.R.; Paroni, R. Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia 2006, 49, 2824–2827.
- Watts, T.; Berti, I.; Sapone, A.; Gerarduzzi, T.; Not, T.; Zielke, R.; Fasano, A. Role of the intestinal tight junction modulator zonulin in the pathogenesis of type I diabetes in BB diabetic-prone rats. Proc. Natl. Acad. Sci. USA 2005, 102, 2916–2921.
- Sapone, A.; de Magistris, L.; Pietzak, M.; Clemente, M.G.; Tripathi, A.; Cucca, F.; Lampis, R.; Kryszak, D.; Carteni, M.; Generoso, M.; et al. Zonulin upregulation is associated with increased gut permeability in subjects with type 1 diabetes and their relatives. Diabetes 2006, 55, 1443–1449.
- Visser, J.T.; Lammers, K.; Hoogendijk, A.; Boer, M.W.; Brugman, S.; Beijer-Liefers, S.; Zandvoort, A.; Harmsen, H.; Welling, G.; Stellaard, F.; et al. Restoration of impaired intestinal barrier function by the hydrolysed casein diet contributes to the prevention of type 1 diabetes in the diabetes-prone BioBreeding rat. Diabetologia 2010, 53, 2621–2628.
- Guo, X.K.; Ou, J.; Liang, S.; Zhou, X.; Hu, X. Epithelial Hes1 maintains gut homeostasis by preventing microbial dysbiosis. Mucosal Immunol. 2018, 11, 716–726.
- Jensen, J.; Pedersen, E.E.; Galante, P.; Hald, J.; Heller, R.S.; Ishibashi, M.; Kageyama, R.; Guillemot, F.; Serup, P.; Madsen, O.D. Control of endodermal endocrine development by Hes-1. Nat. Genet. 2000, 24, 36–44.
- Suzuki, K.; Fukui, H.; Kayahara, T.; Sawada, M.; Seno, H.; Hiai, H.; Kageyama, R.; Okano, H.; Chiba, T. Hes1-deficient mice show precocious differentiation of Paneth cells in the small intestine. Biochem. Biophys. Res. Commun. 2005, 328, 348–352.
- Yang, Q.; Bermingham, N.A.; Finegold, M.J.; Zoghbi, H.Y. Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science 2001, 294, 2155–2158.
- Liu, X.; Lu, J.; Liu, Z.; Zhao, J.; Sun, H.; Wu, N.; Liu, H.; Liu, W.; Hu, Z.; Meng, G.; et al. Intestinal Epithelial Cell-Derived LKB1 Suppresses Colitogenic Microbiota. J. Immunol. 2018, 200, 1889–1900.
- Shimada, T.; Koike, T.; Yamagata, M.; Yoneda, M.; Hiraishi, H. Regulation of TFF3 expression by homeodomain protein CDX2. Regul. Pept. 2007, 140, 81–87.
- Beuling, E.; Baffour-Awuah, N.Y.; Stapleton, K.A.; Aronson, B.E.; Noah, T.K.; Shroyer, N.F.; Duncan, S.A.; Fleet, J.C.; Krasinski, S.D. GATA factors regulate proliferation, differentiation, and gene expression in small intestine of mature mice. Gastroenterology 2011, 140, 1219–1229.e2.
- Beuling, E.; Aronson, B.E.; Tran, L.M.; Stapleton, K.A.; ter Horst, E.N.; Vissers, L.A.; Verzi, M.P.; Krasinski, S.D. GATA6 is required for proliferation, migration, secretory cell maturation, and gene expression in the mature mouse colon. Mol. Cell. Biol. 2012, 32, 3392–3402.
- Laudisi, F.; Stolfi, C.; Bevivino, G.; Maresca, C.; Franze, E.; Troncone, E.; Lolli, E.; Marafini, I.; Pietrucci, D.; Teofani, A.; et al. GATA6 deficiency leads to epithelial barrier dysfunction and enhances susceptibility to gut inflammation. J. Crohn’s Colitis 2021, jjab145.
- Mori-Akiyama, Y.; van den Born, M.; van Es, J.H.; Hamilton, S.R.; Adams, H.P.; Zhang, J.; Clevers, H.; de Crombrugghe, B. SOX9 is required for the differentiation of paneth cells in the intestinal epithelium. Gastroenterology 2007, 133, 539–546.
- Riba, A.; Olier, M.; Lacroix-Lamande, S.; Lencina, C.; Bacquie, V.; Harkat, C.; Gillet, M.; Baron, M.; Sommer, C.; Mallet, V.; et al. Paneth Cell Defects Induce Microbiota Dysbiosis in Mice and Promote Visceral Hypersensitivity. Gastroenterology 2017, 153, 1594–1606.e2.
- Laukoetter, M.G.; Nava, P.; Lee, W.Y.; Severson, E.A.; Capaldo, C.T.; Babbin, B.A.; Williams, I.R.; Koval, M.; Peatman, E.; Campbell, J.A.; et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J. Exp. Med. 2007, 204, 3067–3076.
- Wada, M.; Tamura, A.; Takahashi, N.; Tsukita, S. Loss of claudins 2 and 15 from mice causes defects in paracellular Na+ flow and nutrient transport in gut and leads to death from malnutrition. Gastroenterology 2013, 144, 369–380.
- Odenwald, M.A.; Choi, W.; Kuo, W.T.; Singh, G.; Sailer, A.; Wang, Y.; Shen, L.; Fanning, A.S.; Turner, J.R. The scaffolding protein ZO-1 coordinates actomyosin and epithelial apical specializations in vitro and in vivo. J. Biol. Chem. 2018, 293, 17317–17335.
- Kuo, W.T.; Zuo, L.; Odenwald, M.A.; Madha, S.; Singh, G.; Gurniak, C.B.; Abraham, C.; Turner, J.R. The Tight Junction Protein ZO-1 Is Dispensable for Barrier Function but Critical for Effective Mucosal Repair. Gastroenterology 2021, 161, 1924–1939.
- Marchelletta, R.R.; Krishnan, M.; Spalinger, M.R.; Placone, T.W.; Alvarez, R.; Sayoc-Becerra, A.; Canale, V.; Shawki, A.; Park, Y.S.; Bernts, L.H.; et al. T cell protein tyrosine phosphatase protects intestinal barrier function by restricting epithelial tight junction remodeling. J. Clin. Investig. 2021, 131, e138230.
- Liso, M.; De Santis, S.; Verna, G.; Dicarlo, M.; Calasso, M.; Santino, A.; Gigante, I.; Eri, R.; Raveenthiraraj, S.; Sobolewski, A.; et al. A Specific Mutation in Muc2 Determines Early Dysbiosis in Colitis-Prone Winnie Mice. Inflamm. Bowel Dis. 2020, 26, 546–556.
- Eri, R.D.; Adams, R.J.; Tran, T.V.; Tong, H.; Das, I.; Roche, D.K.; Oancea, I.; Png, C.W.; Jeffery, P.L.; Radford-Smith, G.L.; et al. An intestinal epithelial defect conferring ER stress results in inflammation involving both innate and adaptive immunity. Mucosal Immunol. 2011, 4, 354–364.
- Shroyer, N.F.; Wallis, D.; Venken, K.J.; Bellen, H.J.; Zoghbi, H.Y. Gfi1 functions downstream of Math1 to control intestinal secretory cell subtype allocation and differentiation. Genes Dev. 2005, 19, 2412–2417.
- Chen, Z.; Luo, J.; Li, J.; Kim, G.; Chen, E.S.; Xiao, S.; Snapper, S.B.; Bao, B.; An, D.; Blumberg, R.S.; et al. Foxo1 controls gut homeostasis and commensalism by regulating mucus secretion. J. Exp. Med. 2021, 218, e20210324.
- Simms, L.A.; Doecke, J.D.; Walsh, M.D.; Huang, N.; Fowler, E.V.; Radford-Smith, G.L. Reduced alpha-defensin expression is associated with inflammation and not NOD2 mutation status in ileal Crohn’s disease. Gut 2008, 57, 903–910.
- Wehkamp, J.; Harder, J.; Weichenthal, M.; Schwab, M.; Schaffeler, E.; Schlee, M.; Herrlinger, K.R.; Stallmach, A.; Noack, F.; Fritz, P.; et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut 2004, 53, 1658–1664.
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263.
- Cadwell, K.; Patel, K.K.; Maloney, N.S.; Liu, T.C.; Ng, A.C.; Storer, C.E.; Head, R.D.; Xavier, R.; Stappenbeck, T.S.; Virgin, H.W. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 2010, 141, 1135–1145.
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.J.; Bock, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276.
- Koslowski, M.J.; Kubler, I.; Chamaillard, M.; Schaeffeler, E.; Reinisch, W.; Wang, G.; Beisner, J.; Teml, A.; Peyrin-Biroulet, L.; Winter, S.; et al. Genetic variants of Wnt transcription factor TCF-4 (TCF7L2) putative promoter region are associated with small intestinal Crohn’s disease. PLoS ONE 2009, 4, e4496.
- Wehkamp, J.; Wang, G.; Kubler, I.; Nuding, S.; Gregorieff, A.; Schnabel, A.; Kays, R.J.; Fellermann, K.; Burk, O.; Schwab, M.; et al. The Paneth cell alpha-defensin deficiency of ileal Crohn’s disease is linked to Wnt/Tcf-4. J. Immunol. 2007, 179, 3109–3118.
- Neal, M.D.; Sodhi, C.P.; Jia, H.; Dyer, M.; Egan, C.E.; Yazji, I.; Good, M.; Afrazi, A.; Marino, R.; Slagle, D.; et al. Toll-like receptor 4 is expressed on intestinal stem cells and regulates their proliferation and apoptosis via the p53 up-regulated modulator of apoptosis. J. Biol. Chem. 2012, 287, 37296–37308.
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cezard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603.
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001, 411, 603–606.
- Cooney, R.; Baker, J.; Brain, O.; Danis, B.; Pichulik, T.; Allan, P.; Ferguson, D.J.; Campbell, B.J.; Jewell, D.; Simmons, A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 2010, 16, 90–97.
- Petnicki-Ocwieja, T.; Hrncir, T.; Liu, Y.J.; Biswas, A.; Hudcovic, T.; Tlaskalova-Hogenova, H.; Kobayashi, K.S. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc. Natl. Acad. Sci. USA 2009, 106, 15813–15818.
- Nigro, G.; Rossi, R.; Commere, P.H.; Jay, P.; Sansonetti, P.J. The cytosolic bacterial peptidoglycan sensor Nod2 affords stem cell protection and links microbes to gut epithelial regeneration. Cell Host Microbe 2014, 15, 792–798.
- Grasberger, H.; Magis, A.T.; Sheng, E.; Conomos, M.P.; Zhang, M.; Garzotto, L.S.; Hou, G.; Bishu, S.; Nagao-Kitamoto, H.; El-Zaatari, M.; et al. DUOX2 variants associate with preclinical disturbances in microbiota-immune homeostasis and increased inflammatory bowel disease risk. J. Clin. Investig. 2021, 131, e141676.
- Pircalabioru, G.; Aviello, G.; Kubica, M.; Zhdanov, A.; Paclet, M.H.; Brennan, L.; Hertzberger, R.; Papkovsky, D.; Bourke, B.; Knaus, U.G. Defensive Mutualism Rescues NADPH Oxidase Inactivation in Gut Infection. Cell Host Microbe 2016, 19, 651–663.
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014, 41, 296–310.
- Dring, M.M.; Goulding, C.A.; Trimble, V.I.; Keegan, D.; Ryan, A.W.; Brophy, K.M.; Smyth, C.M.; Keeling, P.W.; O’Donoghue, D.; O’Sullivan, M.; et al. The pregnane X receptor locus is associated with susceptibility to inflammatory bowel disease. Gastroenterology 2006, 130, 341–348.
- Martinez, A.; Marquez, A.; Mendoza, J.; Taxonera, C.; Fernandez-Arquero, M.; Diaz-Rubio, M.; de la Concha, E.G.; Urcelay, E. Role of the PXR gene locus in inflammatory bowel diseases. Inflamm. Bowel Dis. 2007, 13, 1484–1487.
- Alvarado, D.M.; Chen, B.; Iticovici, M.; Thaker, A.I.; Dai, N.; VanDussen, K.L.; Shaikh, N.; Lim, C.K.; Guillemin, G.J.; Tarr, P.I.; et al. Epithelial Indoleamine 2,3-Dioxygenase 1 Modulates Aryl Hydrocarbon Receptor and Notch Signaling to Increase Differentiation of Secretory Cells and Alter Mucus-Associated Microbiota. Gastroenterology 2019, 157, 1093–1108.e11.
- Metidji, A.; Omenetti, S.; Crotta, S.; Li, Y.; Nye, E.; Ross, E.; Li, V.; Maradana, M.R.; Schiering, C.; Stockinger, B. The Environmental Sensor AHR Protects from Inflammatory Damage by Maintaining Intestinal Stem Cell Homeostasis and Barrier Integrity. Immunity 2018, 49, 353–362.e5.
- Yu, M.; Wang, Q.; Ma, Y.; Li, L.; Yu, K.; Zhang, Z.; Chen, G.; Li, X.; Xiao, W.; Xu, P.; et al. Aryl Hydrocarbon Receptor Activation Modulates Intestinal Epithelial Barrier Function by Maintaining Tight Junction Integrity. Int. J. Biol. Sci. 2018, 14, 69–77.
- Catanzaro, J.R.; Strauss, J.D.; Bielecka, A.; Porto, A.F.; Lobo, F.M.; Urban, A.; Schofield, W.B.; Palm, N.W. IgA-deficient humans exhibit gut microbiota dysbiosis despite secretion of compensatory IgM. Sci. Rep. 2019, 9, 13574.
- Johansen, F.E.; Pekna, M.; Norderhaug, I.N.; Haneberg, B.; Hietala, M.A.; Krajci, P.; Betsholtz, C.; Brandtzaeg, P. Absence of epithelial immunoglobulin A transport, with increased mucosal leakiness, in polymeric immunoglobulin receptor/secretory component-deficient mice. J. Exp. Med. 1999, 190, 915–922.
- Shimada, S.; Kawaguchi-Miyashita, M.; Kushiro, A.; Sato, T.; Nanno, M.; Sako, T.; Matsuoka, Y.; Sudo, K.; Tagawa, Y.; Iwakura, Y.; et al. Generation of polymeric immunoglobulin receptor-deficient mouse with marked reduction of secretory IgA. J. Immunol. 1999, 163, 5367–5373.
- Menta, P.L.R.; Andrade, M.E.R.; Leocadio, P.C.L.; Fraga, J.R.; Dias, M.T.S.; Cara, D.C.; Cardoso, V.N.; Borges, L.F.; Capettini, L.S.A.; Aguilar, E.C.; et al. Wheat gluten intake increases the severity of experimental colitis and bacterial translocation by weakening of the proteins of the junctional complex. Br. J. Nutr. 2019, 121, 361–373.
- Johnson, R.J.; Rivard, C.; Lanaspa, M.A.; Otabachian-Smith, S.; Ishimoto, T.; Cicerchi, C.; Cheeke, P.R.; Macintosh, B.; Hess, T. Fructokinase, Fructans, Intestinal Permeability, and Metabolic Syndrome: An Equine Connection? J. Equine Vet. Sci. 2013, 33, 120–126.
- Gulhane, M.; Murray, L.; Lourie, R.; Tong, H.; Sheng, Y.H.; Wang, R.; Kang, A.; Schreiber, V.; Wong, K.Y.; Magor, G.; et al. High Fat Diets Induce Colonic Epithelial Cell Stress and Inflammation that is Reversed by IL-22. Sci. Rep. 2016, 6, 28990.
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 2012, 487, 104–108.
- Ma, T.Y.; Nguyen, D.; Bui, V.; Nguyen, H.; Hoa, N. Ethanol modulation of intestinal epithelial tight junction barrier. Am. J. Physiol. 1999, 276, G965–G974.
- Elamin, E.; Jonkers, D.; Juuti-Uusitalo, K.; van Ijzendoorn, S.; Troost, F.; Duimel, H.; Broers, J.; Verheyen, F.; Dekker, J.; Masclee, A. Effects of ethanol and acetaldehyde on tight junction integrity: In vitro study in a three dimensional intestinal epithelial cell culture model. PLoS ONE 2012, 7, e35008.
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353.e21.
- Sankaran-Walters, S.; Hart, R.; Dills, C. Guardians of the Gut: Enteric Defensins. Front. Microbiol. 2017, 8, 647.
- Shimizu, Y.; Nakamura, K.; Yoshii, A.; Yokoi, Y.; Kikuchi, M.; Shinozaki, R.; Nakamura, S.; Ohira, S.; Sugimoto, R.; Ayabe, T. Paneth cell alpha-defensin misfolding correlates with dysbiosis and ileitis in Crohn’s disease model mice. Life Sci. Alliance 2020, 3, e201900592.
- Zhou, H.; Zhou, S.Y.; Gillilland, M., 3rd; Li, J.Y.; Lee, A.; Gao, J.; Zhang, G.; Xu, X.; Owyang, C. Bile acid toxicity in Paneth cells contributes to gut dysbiosis induced by high-fat feeding. JCI Insight 2020, 5, e138881.
- Liu, T.C.; Kern, J.T.; Jain, U.; Sonnek, N.M.; Xiong, S.; Simpson, K.F.; VanDussen, K.L.; Winkler, E.S.; Haritunians, T.; Malique, A.; et al. Western diet induces Paneth cell defects through microbiome alterations and farnesoid X receptor and type I interferon activation. Cell Host Microbe 2021, 29, 988–1001.e6.
- Brawner, K.M.; Yeramilli, V.A.; Duck, L.W.; Van Der Pol, W.; Smythies, L.E.; Morrow, C.D.; Elson, C.O.; Martin, C.A. Depletion of dietary aryl hydrocarbon receptor ligands alters microbiota composition and function. Sci. Rep. 2019, 9, 14724.
- Schanz, O.; Chijiiwa, R.; Cengiz, S.C.; Majlesain, Y.; Weighardt, H.; Takeyama, H.; Forster, I. Dietary AhR Ligands Regulate AhRR Expression in Intestinal Immune Cells and Intestinal Microbiota Composition. Int. J. Mol. Sci. 2020, 21, 3189.
- Zhang, Y.G.; Wu, S.; Xia, Y.; Sun, J. Salmonella infection upregulates the leaky protein claudin-2 in intestinal epithelial cells. PLoS ONE 2013, 8, e58606.
- Fasano, A.; Baudry, B.; Pumplin, D.W.; Wasserman, S.S.; Tall, B.D.; Ketley, J.M.; Kaper, J.B. Vibrio cholerae produces a second enterotoxin, which affects intestinal tight junctions. Proc. Natl. Acad. Sci. USA 1991, 88, 5242–5246.
- Fasano, A.; Fiorentini, C.; Donelli, G.; Uzzau, S.; Kaper, J.B.; Margaretten, K.; Ding, X.; Guandalini, S.; Comstock, L.; Goldblum, S.E. Zonula occludens toxin modulates tight junctions through protein kinase C-dependent actin reorganization, in vitro. J. Clin. Investig. 1995, 96, 710–720.
- Jakobsson, H.E.; Rodriguez-Pineiro, A.M.; Schutte, A.; Ermund, A.; Boysen, P.; Bemark, M.; Sommer, F.; Backhed, F.; Hansson, G.C.; Johansson, M.E. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. 2015, 16, 164–177.
- Feng, Y.; Huang, Y.; Wang, Y.; Wang, P.; Song, H.; Wang, F. Antibiotics induced intestinal tight junction barrier dysfunction is associated with microbiota dysbiosis, activated NLRP3 inflammasome and autophagy. PLoS ONE 2019, 14, e0218384.
- Sharma, R.; Young, C.; Neu, J. Molecular modulation of intestinal epithelial barrier: Contribution of microbiota. J. Biomed. Biotechnol. 2010, 2010, 305879.
- van der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 2009, 71, 241–260.
- Beumer, J.; Clevers, H. Cell fate specification and differentiation in the adult mammalian intestine. Nat. Rev. Mol. Cell Biol. 2021, 22, 39–53.
- Fre, S.; Huyghe, M.; Mourikis, P.; Robine, S.; Louvard, D.; Artavanis-Tsakonas, S. Notch signals control the fate of immature progenitor cells in the intestine. Nature 2005, 435, 964–968.
- Calon, A.; Gross, I.; Lhermitte, B.; Martin, E.; Beck, F.; Duclos, B.; Kedinger, M.; Duluc, I.; Domon-Dell, C.; Freund, J.N. Different effects of the Cdx1 and Cdx2 homeobox genes in a murine model of intestinal inflammation. Gut 2007, 56, 1688–1695.
- Mesquita, P.; Jonckheere, N.; Almeida, R.; Ducourouble, M.P.; Serpa, J.; Silva, E.; Pigny, P.; Silva, F.S.; Reis, C.; Silberg, D.; et al. Human MUC2 mucin gene is transcriptionally regulated by Cdx homeodomain proteins in gastrointestinal carcinoma cell lines. J. Biol. Chem. 2003, 278, 51549–51556.
- Lorentz, O.; Duluc, I.; Arcangelis, A.D.; Simon-Assmann, P.; Kedinger, M.; Freund, J.N. Key role of the Cdx2 homeobox gene in extracellular matrix-mediated intestinal cell differentiation. J. Cell Biol. 1997, 139, 1553–1565.
- Hinoi, T.; Lucas, P.C.; Kuick, R.; Hanash, S.; Cho, K.R.; Fearon, E.R. CDX2 regulates liver intestine-cadherin expression in normal and malignant colon epithelium and intestinal metaplasia. Gastroenterology 2002, 123, 1565–1577.
- Keller, M.S.; Ezaki, T.; Guo, R.J.; Lynch, J.P. Cdx1 or Cdx2 expression activates E-cadherin-mediated cell-cell adhesion and compaction in human COLO 205 cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G104–G114.
- Morrisey, E.E.; Ip, H.S.; Lu, M.M.; Parmacek, M.S. GATA-6: A zinc finger transcription factor that is expressed in multiple cell lineages derived from lateral mesoderm. Dev. Biol. 1996, 177, 309–322.
- Menard, S.; Cerf-Bensussan, N.; Heyman, M. Multiple facets of intestinal permeability and epithelial handling of dietary antigens. Mucosal Immunol. 2010, 3, 247–259.
- Schneeberger, E.E.; Lynch, R.D. Structure, function, and regulation of cellular tight junctions. Am. J. Physiol. 1992, 262, L647–L661.
- Gumbiner, B.M. Breaking through the tight junction barrier. J. Cell Biol. 1993, 123, 1631–1633.
- Tsukita, S.; Furuse, M. The structure and function of claudins, cell adhesion molecules at tight junctions. Ann. N. Y. Acad. Sci. 2000, 915, 129–135.
- Harhaj, N.S.; Antonetti, D.A. Regulation of tight junctions and loss of barrier function in pathophysiology. Int. J. Biochem. Cell Biol. 2004, 36, 1206–1237.
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Et Biophys. Acta 2008, 1778, 660–669.
- Clemente, M.G.; De Virgiliis, S.; Kang, J.S.; Macatagney, R.; Musu, M.P.; Di Pierro, M.R.; Drago, S.; Congia, M.; Fasano, A. Early effects of gliadin on enterocyte intracellular signalling involved in intestinal barrier function. Gut 2003, 52, 218–223.
- Tripathi, A.; Lammers, K.M.; Goldblum, S.; Shea-Donohue, T.; Netzel-Arnett, S.; Buzza, M.S.; Antalis, T.M.; Vogel, S.N.; Zhao, A.; Yang, S.; et al. Identification of human zonulin, a physiological modulator of tight junctions, as prehaptoglobin-2. Proc. Natl. Acad. Sci. USA 2009, 106, 16799–16804.
- Zeissig, S.; Burgel, N.; Gunzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72.
- Kucharzik, T.; Walsh, S.V.; Chen, J.; Parkos, C.A.; Nusrat, A. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am. J. Pathol. 2001, 159, 2001–2009.
- Dhawan, P.; Ahmad, R.; Chaturvedi, R.; Smith, J.J.; Midha, R.; Mittal, M.K.; Krishnan, M.; Chen, X.; Eschrich, S.; Yeatman, T.J.; et al. Claudin-2 expression increases tumorigenicity of colon cancer cells: Role of epidermal growth factor receptor activation. Oncogene 2011, 30, 3234–3247.
- Damci, T.; Nuhoglu, I.; Devranoglu, G.; Osar, Z.; Demir, M.; Ilkova, H. Increased intestinal permeability as a cause of fluctuating postprandial blood glucose levels in Type 1 diabetic patients. Eur. J. Clin. Investig. 2003, 33, 397–401.
- Carratu, R.; Secondulfo, M.; de Magistris, L.; Iafusco, D.; Urio, A.; Carbone, M.G.; Pontoni, G.; Carteni, M.; Prisco, F. Altered intestinal permeability to mannitol in diabetes mellitus type I. J. Pediatric Gastroenterol. Nutr. 1999, 28, 264–269.
- Khoshbin, K.; Camilleri, M. Effects of dietary components on intestinal permeability in health and disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G589–G608.
- Johansson, M.E.; Sjovall, H.; Hansson, G.C. The gastrointestinal mucus system in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 352–361.
- Johansson, M.E.; Ambort, D.; Pelaseyed, T.; Schutte, A.; Gustafsson, J.K.; Ermund, A.; Subramani, D.B.; Holmen-Larsson, J.M.; Thomsson, K.A.; Bergstrom, J.H.; et al. Composition and functional role of the mucus layers in the intestine. Cell. Mol. Life Sci. CMLS 2011, 68, 3635–3641.
- Arike, L.; Hansson, G.C. The Densely O-Glycosylated MUC2 Mucin Protects the Intestine and Provides Food for the Commensal Bacteria. J. Mol. Biol. 2016, 428, 3221–3229.
More
Information
Subjects:
Gastroenterology & Hepatology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.7K
Entry Collection:
Gastrointestinal Disease
Revisions:
2 times
(View History)
Update Date:
17 Feb 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No