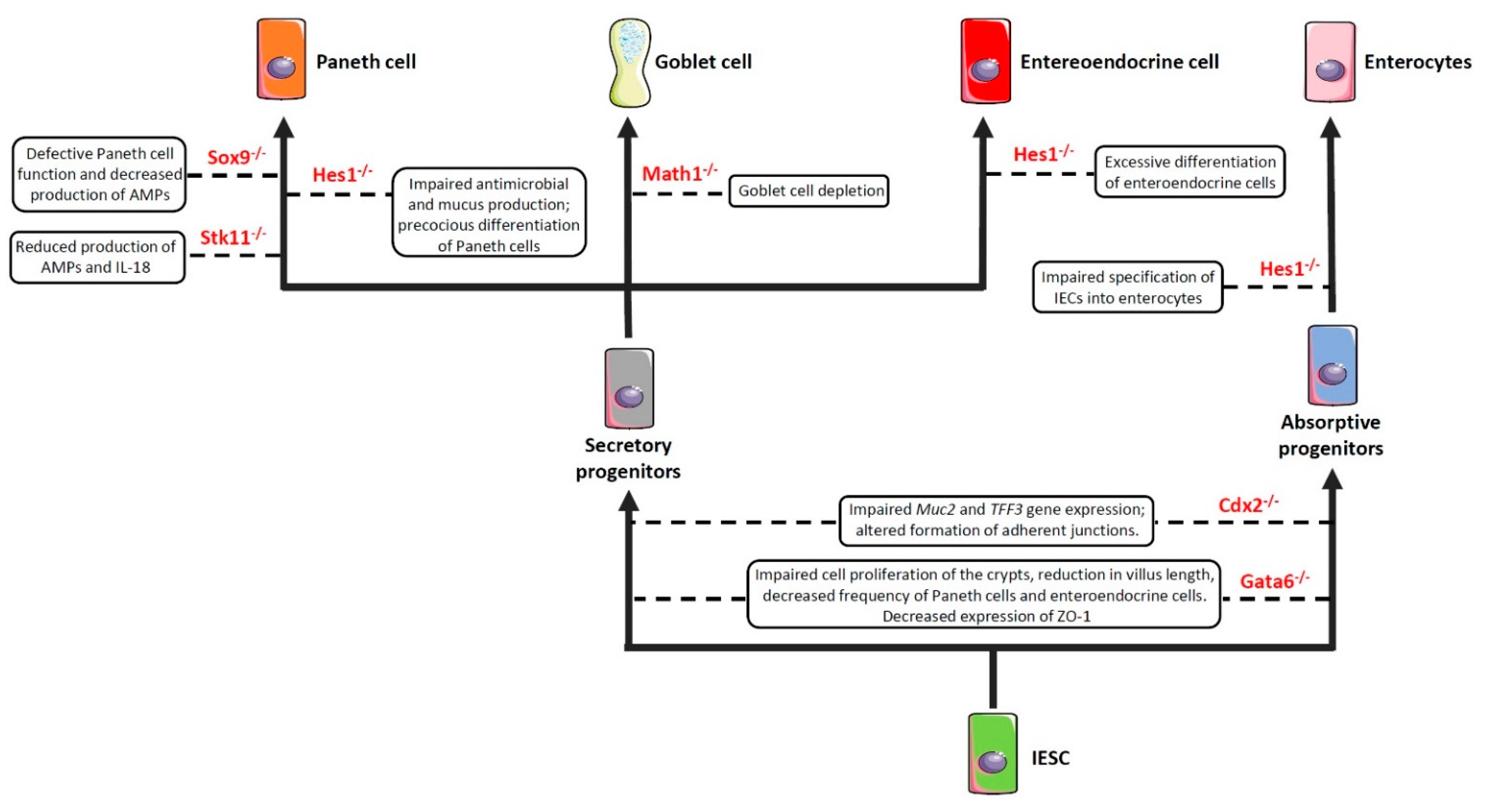
2.1. Impairment of Cell Commitment
As previously anticipated, the intestinal barrier is characterized by a self-renewing epithelium, organized in crypts and villi, including both stem cells and differentiated cells. As the epithelial barrier has to deal with multiple physiologic activities, it requires different specialized cells, some of which are able to produce and secrete several molecules—such as antimicrobial peptides, mucins and hormones—and others that are able to adsorb water and nutrients. To achieve this goal, stem cells, after multiple transit-amplifying (TA) divisions, terminally differentiate into either secretory or absorptive lineages depending on the tightly regulated expression/inhibition of specific transcription factors
[114][115]. For instance, the cell surface receptor Notch drives the cell commitment process by binding to the Notch ligands Deltalike (Dll) and Jagged families
[116]. A cell expressing the Notch ligands will differentiate into a secretory cell (e.g., goblet cell, Paneth cell) upon expression of the
Atonal BHLH Transcription Factor 1 (Atoh1), whereas a neighbour cell expressing the activated Notch receptor will induce the expression of the target gene
hairy and enhancer of split 1 (Hes1)—a Atoh1 inhibitor—and will differentiate into an absorptive cell (e.g., enterocyte)
[54]. Obviously, a dysregulated expression of the above-mentioned proteins definitely compromises the intestinal epithelial cell commitment and, in turn, the intestinal barrier integrity and function, as demonstrated by the association of several polymorphisms in genes encoding commitment-related transcription factors with intestinal barrier dysfunctions and intestinal inflammatory diseases (
Figure 2)
[51][52][53][54][55]. In this context, by employing genetically engineered mouse models, Guo and colleagues demonstrated that deficiency of the
Hes1 gene in IECs negatively affected antimicrobial peptides and mucus production, thus resulting in gut dysbiosis and inflammation
[51]. Along the same line was the demonstration that mice deficient for
Math1 (also referred to as
Atoh1) displayed complete abrogation of goblet cells in the intestine
[54]. Similar results were observed in mice with conditional deletion of the
serine threonine kinase 11 (
Stk11) gene, involved in the differentiation of stem cells into the secretory lineage cell types (e.g., goblet cells and Paneth cells), in IECs
[55]. These animals displayed increased susceptibility to gut inflammation in association with reduced production of antimicrobial peptides and IL-18, as well as an uncontrolled expansion of colitogenic bacteria
[55].
Figure 2. Impaired expression of genes encoding commitment-related transcription factors compromises epithelial cell differentiation and intestinal barrier function. Boxes enclose the effect/s of the knockdown of the genes depicted in red on the indicated cell commitment. Abbreviations: Sox9: SRY-Box Transcription Factor 9; Hes1: Hairy and enhancer of split 1; Stk11: Serine threonine kinase 11; Math1: Mouse atonal homolog 1; Cdx2: Caudal type homeobox 2; Gata6: GATA binding factor 6; Muc2: Mucin 2; TFF3: Trefoil factor 3; ZO-1: Zonula Occludens-1; AMPs: Antimicrobial peptides; IESC: Intestinal epithelial stem cells.
Another important transcription factor involved in the commitment of IECs is the
caudal type homeobox 2 (Cdx2) gene
[117]. Indeed, CDX2 is a positive regulator of the
Muc2 and
Trefoil Factor 3 (TFF3) genes
[56][118] involved in the production and stabilization of the mucus layer, respectively, and whose deficiency induces hypersensitivity to chemically-induced colitis (such as that induced by dextran sulfate sodium (DSS))
[15][56]. In addition, CDX2 controls cell-cell interactions and the expression of cadherins, which are important in the formation of the adherens junctions
[119][120][121]. In support of this view is the evidence that mice bearing one non-functional
Cdx2 allele (
Cdx2+/− mice) displayed increased intestinal permeability and were more susceptible to the abrasive effect of DSS
[117].
The GATA binding factor 6 (GATA6) is a zinc finger transcription factor that regulates cell proliferation, differentiation, and gene expression in several tissues
[122]. For instance, GATA6 is involved in cell proliferation and differentiation along the gastrointestinal tract
[57][58]. In particular, conditional deletion of
Gata6 in IECs resulted in impaired cell proliferation of the crypts, reduction in villus length, decreased frequency of Paneth cells and enteroendocrine cells, increased number of goblet-like cells, and dysregulated expression of enterocyte-related genes in the ileum
[57]. Similar alterations were observed in the colon, where
Gata6 deficiency affected stem cell proliferation and differentiation into Paneth cells, enteroendocrine cells, and enterocytes
[58]. Researchers' study has recently demonstrated that conditional deletion of
Gata6 in the gut epithelium significantly affected intestinal barrier integrity, leading to decreased expression of the tight junction-related protein zonula occludens-1 (ZO-1), and resulting in increased paracellular permeability, microbial dysbiosis, and susceptibility to gut inflammation
[59]. Interestingly, researchers also reported a decreased expression of GATA6 in the intestinal epithelium of IBD patients, thus suggesting that a reduced expression of this transcription factor may contribute to intestinal barrier dysfunction in these subjects
[59]. In the intestinal epithelium, defects in Paneth cell function—and the consequent decrease in the antimicrobial peptide production—may also result from the deletion of
Sox9 [60]. Indeed, by generating mice that harbored a conditional
Sox9 gene and a Villin-Cre transgene, Mori-Akiyama et al. reported that lack of
Sox9 expression in the intestinal epithelium of
Sox9fl/fl Villin-Cre
+ mice resulted in the complete absence of differentiated Paneth cells, although the differentiation of other intestinal epithelial cell subsets (e.g., goblet cells, enterocytes) was not affected. Moreover,
Sox9 deficiency also lead to crypt enlargement, a marked increase in cell proliferation throughout the crypts, as well as a replacement of the Paneth cells by proliferating epithelial cells
[60]. More recently, by employing the same conditional mouse model, Riba and colleagues showed that
Sox9 deletion in the intestinal epithelium reduced lysozyme production. This effect resulted in significant microbial dysbiosis, characterized by
E. coli overgrowth and ultimately leading to visceral hypersensitivity
[61].
2.2. Impairment of Epithelial Junctional Complexes
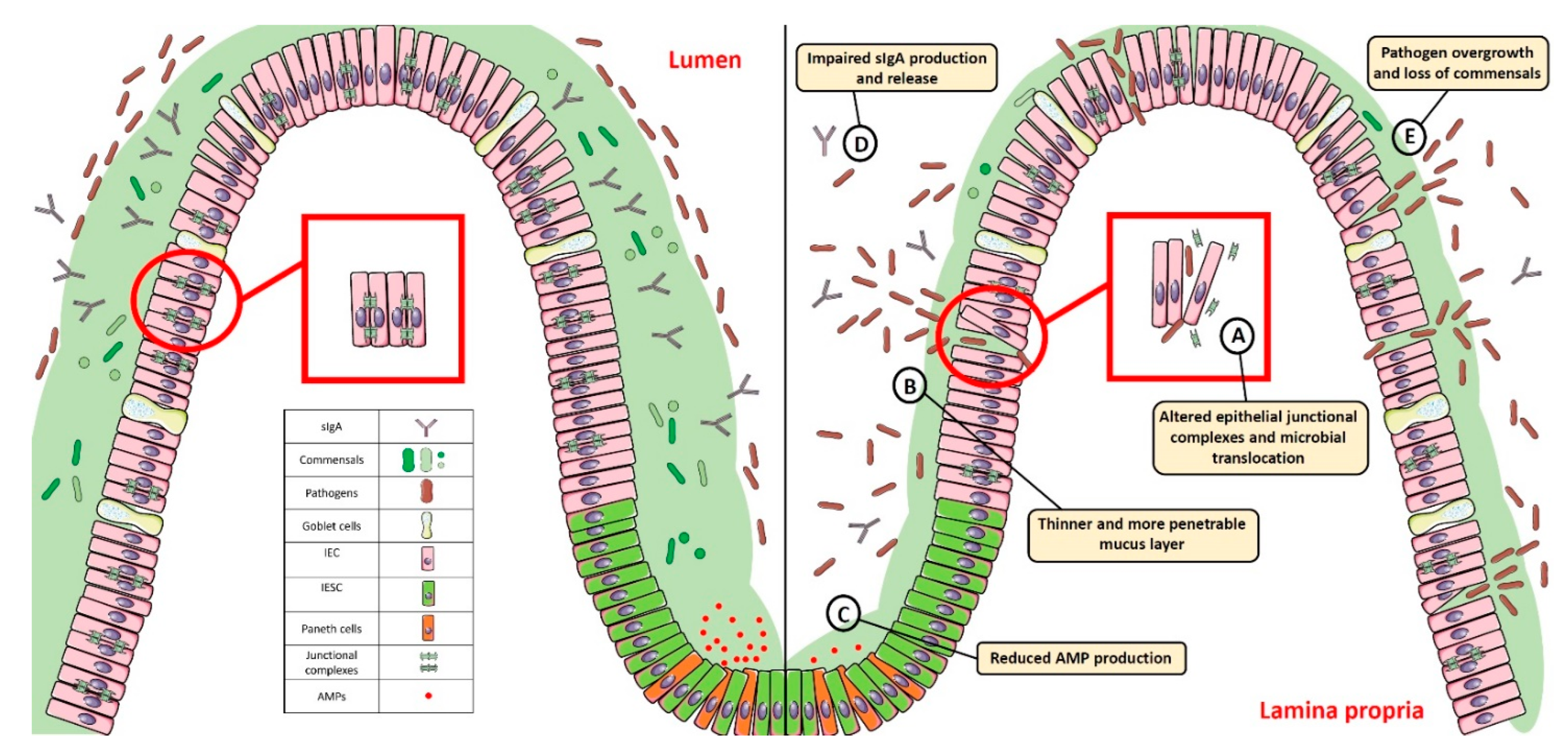
The intestinal epithelial barrier’s main function is to protect the host from luminal antigens, pathogens, and toxins, while allowing selective permeability to water, nutrients, and electrolytes. In particular, transcellular permeability, involved in solute transport through the epithelial cells, is mediated by selective transporters for amino acids, electrolytes, short chain fatty acids, and sugars
[123]. Paracellular permeability, instead, occurs through intercellular junctional complexes encompassing adherens junctions (AJs), tight junctions (TJs), and desmosomes
[124][125]. These transmembrane proteins, localized both at the apical-lateral membrane junction and along the lateral membrane, mediate the contact between adjacent IECs, thus sealing the intracellular spaces. The AJs (e.g., catenins, cadherins) and desmosomes (e.g., desmoglein, desmocollins) regulate the mechanical linkage of adjacent cells, while the TJs (e.g., ZO-1, claudin-2, occludins, Junctional Adhesion Molecule) form an apical junctional complex that seals the intercellular space and modulates selective paracellular permeability
[124][125][126][127][128].
Alterations in the formation/distribution of the intercellular junctional complexes, which may occur in men with specific genetic susceptibilities, as well as in response to dietary factors and bacterial infections, may result in intestinal epithelial barrier breakdown and translocation of the luminal content into the lamina propria, leading to gut dysbiosis, uncontrolled immune/inflammatory responses, and, ultimately, pathological conditions
[10].
For instance, gliadin (a glycoprotein representing the major component of wheat gluten) has been reported to deeply affect the expression and distribution of several junctional complexes in the small intestine of celiac patients by binding to CXC motif receptor 3 (CXCR3) on epithelial cells
[20]. This interaction induces the release of zonulin, a human protein analogue of the Zonula occludens toxin (ZOT) from
Vibrio cholerae, through the recruitment of Myeloid differentiation primary response (MyD)-88
[20][21][129][130]. Increased levels of zonulin were detected in the intestinal tissues taken from celiac disease patients during the acute phase compared to those taken from healthy controls. Once released, Zonulin leads to transactivation of EGF receptor (EGFR) via proteinase-activated receptor 2 (PAR2) activation in the intestinal epithelium and subsequent tight junction disassembly
[20][21][129][130].
The impairment of the epithelial junctional complexes importantly contributes to the development of other chronic inflammatory conditions, such as IBD. For example, increased expression of claudin-2, as well as impaired expression and redistribution of claudin-5, -8 and occludin were reported in Crohn’s disease patients, leading to increased intestinal permeability and bacterial translocation
[131]. A similar severe condition was described also in the colonic mucosa of patients with ulcerative colitis, in association with the dysregulated expression of occludin, ZO-1, claudin-1, JAM, beta-catenin, E-cadherin, and the consequent transepithelial migration of neutrophils
[132].
The above-mentioned chronic inflammatory conditions importantly contribute to the development and progress of colorectal carcinogenesis. Interestingly, increased expression of claudin-1 and claudin-2 was found to correlate with inflammatory activity, IBD-associated dysplasia, and sporadic adenomas
[36]. Similarly, Dhawan et al. observed that claudin-1 expression was increased in colon carcinomas and metastatic lesions and played a key role for tumorigenesis and invasiveness of colonic epithelial cells
[35]. Claudin-2 was also reported to be increased in tissues taken from CRC and IBD-associated CRC patients and to promote and sustain cell proliferation and tumor growth in cultured cells and experimental models
[133].
Dysfunctions of the epithelial junctional complexes and the consequent increase of intestinal permeability and gut dysbiosis correlate with the development and progression of other pathological conditions. In particular, increased intestinal permeability was seen to precede and/or to be an early biomarker of diabetes development in patients, as well as in experimental models of the disease
[45][134][135]. Moreover, increased serum levels of zonulin, in association with altered intestinal permeability, were described in a subgroup of patients with type 1 diabetes and their first-degree relatives, suggesting this molecule as a valid early biomarker of disease development
[49].
Animal models employing genetically engineered mice have helped to better understand the link between junctional complex dysregulation and the development of dysbiosis and pathologic conditions. In this context, Laukoetter et al. reported a role for Junctional Adhesion Molecule (JAM)-A, a TJ component contributing to the control of barrier function and leukocyte migration, in regulating intestinal permeability and inflammation in vivo
[62]. Indeed, despite showing normal epithelial architecture,
JAM-A knockout mice developed low-grade colonic inflammation (characterized by enhanced polymorphonuclear leukocyte infiltration and large lymphoid aggregates not seen in sham mice)
[62]. Barrier function experiments revealed increased mucosal permeability, as indicated by enhanced dextran flux, and decreased transepithelial electrical resistance in
JAM-A knockout mice compared to wild-type control mice
[62]. Consistently,
JAM-A deficiency increased the permeability of in vitro monolayers derived from the human colonic epithelial cell line SK-CO15 compared with control. Moreover,
JAM-A deficient mice were more susceptible to the DSS-driven experimental colitis compared to controls, although the colonic mucosa showed less injury and increased epithelial proliferation
[62]. Analyses of other TJ-related proteins showed increased expression of claudin-10 and -15, both of which tune TJ barrier function by the formation of ion-selective pores, following
JAM-A knockdown in the colonic mucosa of mice and in SK-CO15 cell monolayers
[62].
In a later article, Wada and colleagues reported that mice with the double knockdown of
claudin-2 (Cldn-2) and
claudin-15 (Cldn-15) genes had impaired paracellular Na
+ flow and subsequent malnutrition, leading to infant death
[63].
By employing cultured epithelial cells and an intestinal epithelial-specific knockout mouse (that is,
Tjp1fl/fl Villin-Cre
+ mouse), Odenwald and co-workers showed that the TJ scaffolding protein ZO-1 was essential for development of unified apical surfaces in vitro and in vivo. In detail, conditional deletion of ZO-1 in IECs of
Tjp1fl/fl Villin-Cre
+ mice did not significantly alter crypt-villus architecture, whereas it affected apical tissue continuity, which is by characterized apical surface brush border membrane, and the presence of crevasses at intercellular junctions between enterocytes, likely by modulating actomyosin contraction and membrane traffic
[64]. Recently, Kuo and colleagues reported decreased ZO-1 expression, both at RNA and protein level, in intestinal mucosal biopsies isolated from IBD patients as compared with those isolated from healthy controls
[65]. Loss of ZO-1 expression in epithelial cells in
Tjp1fl/fl Villin-Cre
+ mice did not promote spontaneous disease, but it exacerbated tissue damage and weight loss during experimental colitis, as well as delayed the mucosal healing
[65]. The authors also reported that ZO-1 is critically involved in the cell division phase upon damage. In particular, by associating with the centriole and mitotic spindle, ZO-1 contributed to both Wnt–β-catenin signaling and mitotic spindle orientation, suggesting that ZO-1 may actively contribute to the intestinal epithelial barrier restoration
[65]. In line with these observations, researchers recently found that loss of
Gata6 expression in IECs of genetically engineered mice resulted in increased intestinal permeability, gut dysbiosis, and microbial-driven intestinal inflammation. These effects were associated with decreased ZO-1 expression and epithelial damage both in the ileum and colon. Experiments in cultured cells suggested that ZO-1 expression could be directly modulated by GATA6
[59]. Recently, Marchelletta and colleagues reported that the impaired function of T cell protein tyrosine phosphatase (TCPTP), encoded by the
protein tyrosine phosphatase non-receptor type 2 (PTPN2) gene, contributed to the epithelial tight junction protein remodeling and increased intestinal permeability
[66]. In particular,
Tcptp-deficient mice showed increased claudin-2 expression, intestinal permeability, and inflammatory cytokine production
[66]. In detail, TCPTP was able to maintain the localization of ZO-1 and occludin at apical tight junctions, as well as to modulate the turn-over of claudin-2, a cation pore-forming transmembrane protein, by upregulating the serine metalloproteinase matriptase, which promoted claudin-2 proteosomal degradation
[66].
Apart from defects in the above-mentioned epithelial junction-related molecules, several dietary factors may contribute to increase intestinal permeability and trigger/amplify pathologic conditions
[136]. A good example in this regard is given by gluten, which, in addition to its well-known detrimental effects on barrier integrity and TJ protein activity in celiac disease, can actively promote dysregulation of intestinal barrier function in non-celiac patients. Of note, mice exposed to a gluten-rich diet showed alterations in adherent junctions and desmosomes, resulting in increased intestinal permeability and susceptibility to DSS-driven experimental colitis
[95].
Glucose and fructose are additional macronutrients found to trigger TJ and AJ protein dysfunction, thus promoting changes in microbiota composition, increased susceptibility to pathogen infection, as well as metabolic syndrome
[44]. In mouse experimental models, uncontrolled metabolism of fructose in the liver and in the small intestine, due to the excessive delivery of this sugar (15% in water for 3 weeks), induced the transcriptional expression of fructokinase (a protein involved in fructose metabolism), TJ alterations, energy depletion, oxidative stress, and chronic inflammation
[96]. On the other hand, mice deficient of the fructokinase isoforms A and C (
KHK-A, KHK-C) were protected from such detrimental effects. Notably, loss of KHK-A function only did not prevent alterations in TJs, thus suggesting that intestinal barrier impairment was mainly mediated by KHK-C activity
[96].
Detrimental effects of dietary fats on the epithelial junctional complexes have been also reported by several studies. In particular, mice exposed to a high-fat diet for 3, 11, and 22 weeks showed induction of endoplasmic reticulum (ER) stress in IECs, as well as an impairment of Claudin-1 expression and mucus barrier, with the consequent increase of endotoxin serum levels and gut dysbiosis
[97]. Similarly, Devkota and colleagues demonstrated that the increased availability of taurocholic bile acid, due to the consumption of a diet high in saturated (milk derived)-fat, promoted the expansion of the low abundance pathobiont
Bilophila wadsworthia (a member of the
Deltaproteobacteria), which, in turn, was able to impair intestinal barrier integrity in genetically susceptible
Il-10−/− mice due to its sulphite-reducing activity
[98]. Another dietary habit found to affect TJ activity is ethanol consumption. Exposure to non-cytotoxic doses of ethanol (as those detected in the blood of moderate drinkers) impaired paracellular permeability in vitro due to alterations in ZO-1 and occludin localization
[99][100].
Both localization and activity of epithelial junctional complexes can also be affected by pathogen invasion and toxin secretion. For instance,
Salmonella typhimurium was found to up-regulate the colonic expression of the leaky protein claudin-2, which plays an opposite role in the modulation of intestinal permeability compared to other TJ proteins involved in barrier maintenance, thus facilitating bacterial invasion
[108].
Vibrio cholerae, instead, was reported to target the intestinal epithelial barrier by producing the zonula occludens toxin (ZOT), which transiently affects the paracellular permeability in the small intestine by opening TJs through a protein kinase C-dependent actin reorganization
[109][110].
On its side, antibiotic treatment dramatically influences intestinal permeability by compromising host microbial ecology. In particular, mice exposed to antibiotics for 2 weeks developed mucosal dysbiosis characterized by decreased production of short-chain fatty acids, such as butyrate (known to sustain barrier function and integrity), by commensals
[112]. Moreover, antibiotic treatment hampered intestinal TJ function and increased intestinal permeability by reducing the expression of ZO-1, occluding, and claudin-1
[112]. Similar results were obtained in antibiotic-treated germ-free mice, which presented altered microvilli morphology and reduced rate of intestinal epithelial cell turnover compared to sham mice
[113]. Altogether, these results indicate a key role for commensal microbiota in preserving epithelial junctional complexes and gut barrier integrity, highlighting a possible detrimental effect of antibiotic exposure on such a fine balance.
2.3. Thinning/Depletion of the Mucus Layer
Goblet cells are specialized IECs able to synthetize and secrete mucin proteins into the lumen. Mucin proteins are pivotal in creating a protective mucus layer acting against pathogens, chemicals, and mechanical stress in order to maintain gut homeostasis and protect the inner mucosal surface
[137]. The mucus layer, mainly composed of water, electrolytes, lipids, and glycosylated mucins
[138], represents an important source of antimicrobial peptides and immunoglobulins and can directly interact with commensals, providing nutrients and attachment sites depending on the mucin glycosylation profile
[139].
Mucolytic bacteria (e.g.,
Akkermansia muciniphila, Bacteroides thetaiotaomicron, Ruminococcus gnavus, Ruminococcus torques) represent an important class of commensals as they are able to digest glycans (from dietary fibers) and mucins through glycosidase enzymes, and to produce, in turn, short chain fatty acids (such as acetate and butyrate) acting as energy source for colonocytes and contributing to protect the intestinal barrier integrity
[139]. However, the fine balance between goblet cell-mediated replenishment of mucus and its degradation by commensals can be affected by a fiber-deprived diet, as indicated by the fact that mice subjected to intermittent dietary fiber deprivation presented a thinner mucus layer due to O-linked glycan digestion by the fiber-deprived microbiota
[101]. Thus, enrichment in mucus-degrading bacteria may impair the mucus layer thickness and viscosity and promote enteric pathogens adherence and penetration, ultimately causing gut dysbiosis and chronic intestinal inflammation
[67][101]. These pathological alterations were observed in the
Winnie murine model of spontaneous colitis, characterized by a missense mutation in the
Muc2 gene
[16][67]. The phenotype of
Winnie mice was characterized by altered mucus production as early as 4 weeks of age, with ensuing intestinal barrier dysfunction, gut dysbiosis, and inflammation
[16][67]. In particular, impaired
Muc2 expression affected the number of goblet cells, which underwent unresolved ER stress and accumulation of mucin precursors
[16][67]. All these processes were associated with apoptotic cell death, increased intestinal permeability, pathogen penetration into the inner mucus layer, and adherence to epithelial cells, as well as bacterial translocation into the lamina propria
[16][67]. The subsequent uncontrolled immune response towards pathogens (e.g., enhanced dendritic cell activation, T-helper cytokine production) promoted chronic intestinal inflammation and gut dysbiosis, characterized by the outgrowth of
Bacteroidetes and
Verrucomicrobia (such as
Akkermansia muciniphila)
[67][68].
Impaired mucus layer integrity can also depend on mutations in the
Gfi1 gene. Gfi1 functions downstream of
Math1 in the intestinal epithelium and encodes molecules involved in the stem cell differentiation into the different secretory cell lineages
[69]. In particular,
Gfi1-deficient mice displayed alterations in terminal differentiation and morphology of goblet cells and Paneth cells, together with accumulation of immature secretory progenitors, as well as a decrease in mucin and antimicrobial peptide release
[69]. Recently, the Foxo1 trascription factor was described to be critically involved in mucin granule release through autophagy
[70]. In particular,
Foxo1fl/fl Villin-Cre
+ mice showed impaired mucus layer formation and subsequent dysbiosis, resulting in disrupted intestinal barrier integrity and enhanced susceptibility to infection and tissue inflammation. Moreover,
Foxo1 deficiency in IECs resulted in the overgrowth of mucin-degrading bacteria and a decrease of short-chain fatty acid-producing microbial species, which further affected the intestinal barrier function
[70].
In addition to mucus production and degradation, gut microbiota composition is able to influence the mucus properties. In this regard, Jakobsson and colleagues reported that the mucus layer of germ-free mice was characterized by a higher mucus penetrability as compared to conventional mice
[111]. Moreover, mice with identical genetic background, but hosted in two rooms of the same specific pathogen-free animal facility, showed different mucus properties, evidenced by the fact that one colony had an impenetrable inner mucus layer, whereas the other showed opposite features
[111]. The authors suggested that these differences relied on changes in the gut microbiota composition as the different mucus phenotypes were acquired by germ-free mice upon faecal microbiota transplantation
[111]. In particular, mice with an impenetrable inner mucus layer showed increased frequency of the
Erysipelotrichi class, whereas
Proteobacteria and
TM7 expanded in mice with more penetrable mucus
[111]. Hence, even genetically identical animals housed in the same facility may have distinct microbiotas and barrier structures
[111].
Taken together, these results highlight the mutualistic effects between the gut microbial community and the mucus layer and their consequences on intestinal barrier integrity and function.