 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Consolato Sergi | + 1958 word(s) | 1958 | 2022-02-10 04:52:38 | | | |
| 2 | Dean Liu | -21 word(s) | 1937 | 2022-02-11 01:57:36 | | |
Video Upload Options
The term biliary atresia has substituted the original term of “extrahepatic biliary atresia”, which has been in use for several generations. The concept was related to the often-identified absence of gallbladder with an obliterated cord at the site the extrahepatic biliary system. It is now known that biliary atresia is a necro-inflammatory and fibro-obliterative process of both intrahepatic and extrahepatic biliary tract.
1. Introduction
Despite the fact that the etiology is far from being known, pediatric surgeries conducted last century worldwide were incredibly supportive for the clarification of the nosology of this disease [1][2][3]. Mainly, Morio Kasai, a Japanese pediatric surgeon, may be considered the pioneer of a specific pediatric surgery that may be offered to parents of a child with biliary atresia. His name is indelibly bound to the palliative surgical procedure he ideated in Japan [4]. The porto-enterostomy is indeed the major palliative procedure, which may be implemented before a liver is available for transplantation (orthotopic transplant or transplant of a liver from a recently deceased donor, a living donor transplant, or a split type of liver transplant) can be put forward. The biliary atresia is a very complex disease, not just complicated, and several genes have been investigated thoroughly. Some of these genes may be highly relevant to this disease, although their role is mostly not known.
2. Pathological Anatomy of the Biliary Atresia
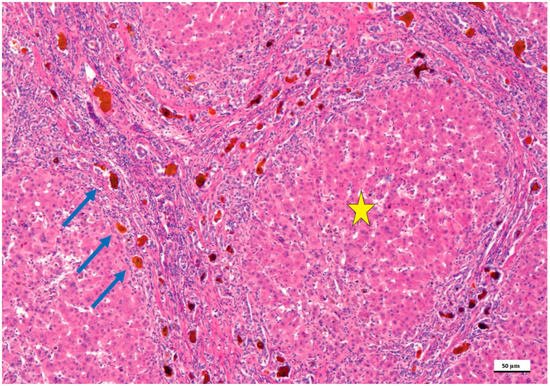
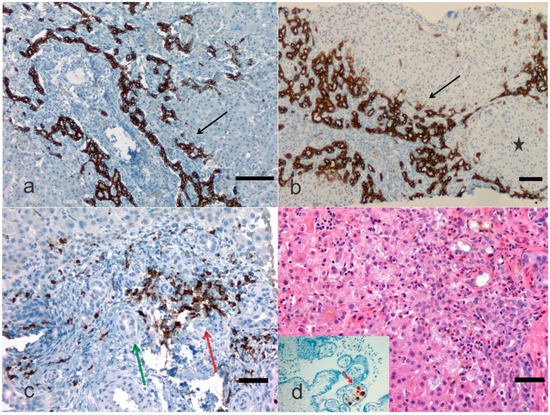
3. Treatment
References
- Pang, W.B.; Zhang, T.C.; Chen, Y.J.; Peng, C.H.; Wang, Z.M.; Wu, D.Y.; Wang, K. Ten-Year Experience in the Prevention of Post-Kasai Cholangitis. Surg. Infect. 2019, 20, 231–235.
- Chan, K.W.E.; Lee, K.H.; Wong, H.Y.V.; Tsui, S.Y.B.; Mou, J.W.C.; Tam, Y.H.P. Ten-Year Native Liver Survival Rate After Laparoscopic and Open Kasai Portoenterostomy for Biliary Atresia. J. Laparoendosc. Adv. Surg. Tech. A 2019, 29, 121–125.
- Wong, Z.H.; Davenport, M. What Happens after Kasai for Biliary Atresia? A European Multicenter Survey. Eur. J. Pediatr. Surg. 2019, 29, 1–6.
- Garcia, A.V.; Cowles, R.A.; Kato, T.; Hardy, M.A. Morio Kasai: A remarkable impact beyond the Kasai procedure. J. Pediatr. Surg. 2012, 47, 1023–1027.
- Sergi, C.; Benstz, J.; Feist, D.; Nutzenadel, W.; Otto, H.F.; Hofmann, W.J. Bile duct to portal space ratio and ductal plate remnants in liver disease of infants aged less than 1 year. Pathology 2008, 40, 260–267.
- Dorn, L.; Menezes, L.F.; Mikuz, G.; Otto, H.F.; Onuchic, L.F.; Sergi, C. Immunohistochemical detection of polyductin and co-localization with liver progenitor cell markers during normal and abnormal development of the intrahepatic biliary system and in adult hepatobiliary carcinomas. J. Cell. Mol. Med. 2009, 13, 1279–1290.
- Sergi, C.M. Genetics of Biliary Atresia: A Work in Progress for a Disease with an Unavoidable Sequela into Liver Cirrhosis following Failure of Hepatic Portoenterostomy. In Liver Cirrhosis—Debates and Current Challenges; Tsoulfas, G., Ed.; IntechOpen: London, UK, 2019.
- Carvalho, N.M.N.; Torres, S.M.; Cavalcante, J.C.B.; Ximenes, A.C.M.; Landim Junior, J.A.; Moreira, S. Hepatoportoenterostomy Surgery Technique. J. Pediatr. Surg. 2018, 54, 1715–1718.
- Shimadera, S.; Iwai, N.; Deguchi, E.; Kimura, O.; Ono, S.; Fumino, S.; Higuchi, K. Significance of ductal plate malformation in the postoperative clinical course of biliary atresia. J. Pediatr. Surg. 2008, 43, 304–307.
- Sergi, C.; Adam, S.; Kahl, P.; Otto, H.F. Study of the malformation of ductal plate of the liver in Meckel syndrome and review of other syndromes presenting with this anomaly. Pediatr. Dev. Pathol. 2000, 3, 568–583.
- Russo, P.; Magee, J.C.; Anders, R.A.; Bove, K.E.; Chung, C.; Cummings, O.W.; Finegold, M.J.; Finn, L.S.; Kim, G.E.; Lovell, M.A.; et al. Key Histopathologic Features of Liver Biopsies That Distinguish Biliary Atresia From Other Causes of Infantile Cholestasis and Their Correlation With Outcome: A Multicenter Study. Am. J. Surg. Pathol. 2016, 40, 1601–1615.
- Ihn, K.; Na, Y.; Ho, I.G.; Lee, D.; Koh, H.; Han, S.J. A periodic comparison of the survival and prognostic factors of biliary atresia after Kasai portoenterostomy: A single-center study in Korea. Pediatr. Surg. Int. 2019, 35, 285–292.
- Parolini, F.; Boroni, G.; Milianti, S.; Tonegatti, L.; Armellini, A.; Garcia Magne, M.; Pedersini, P.; Torri, F.; Orizio, P.; Benvenuti, S.; et al. Biliary atresia: 20–40-year follow-up with native liver in an Italian centre. J. Pediatr. Surg. 2018, 54, 1440–1444.
- Hasan, M.S.; Karim, A.B.; Rukunuzzaman, M.; Haque, A.; Akhter, M.A.; Shoma, U.K.; Yasmin, F.; Rahman, M.A. Role of Liver Biopsy in the Diagnosis of Neonatal Cholestasis due to Biliary Atresia. Mymensingh Med. J. 2018, 27, 826–833.
- Madadi-Sanjani, O.; Kuebler, J.F.; Dippel, S.; Gigina, A.; Falk, C.S.; Vieten, G.; Petersen, C.; Klemann, C. Long-term outcome and necessity of liver transplantation in infants with biliary atresia are independent of cytokine milieu in native liver and serum. Cytokine 2018, 111, 382–388.
- Li, S.; Ma, N.; Meng, X.; Zhang, W.; Sun, C.; Dong, C.; Wang, K.; Wu, B.; Gao, W. The effects of Kasai procedure on living donor liver transplantation for children with biliary atresia. J. Pediatr. Surg. 2018, 54, 1436–1439.
- Witt, M.; van Wessel, D.B.E.; de Kleine, R.H.J.; Bruggink, J.L.M.; Hulscher, J.B.F.; Verkade, H.J.; Netherlands Study Group on Biliary Atresia Registry. Prognosis of Biliary Atresia After 2-year Survival With Native Liver: A Nationwide Cohort Analysis. J. Pediatr. Gastroenterol. Nutr. 2018, 67, 689–694.
- Arafa, R.S.; Abdel Haie, O.M.; El-Azab, D.S.; Abdel-Rahman, A.M.; Sira, M.M. Significant hepatic expression of IL-2 and IL-8 in biliary atresia compared with other neonatal cholestatic disorders. Cytokine 2016, 79, 59–65.
- Santos, J.L.; Kieling, C.O.; Meurer, L.; Vieira, S.; Ferreira, C.T.; Lorentz, A.; Silveira, T.R. The extent of biliary proliferation in liver biopsies from patients with biliary atresia at portoenterostomy is associated with the postoperative prognosis. J. Pediatr. Surg. 2009, 44, 695–701.
- Obayashi, J.; Kawaguchi, K.; Manabe, S.; Nagae, H.; Wakisaka, M.; Koike, J.; Takagi, M.; Kitagawa, H. Prognostic factors indicating survival with native liver after Kasai procedure for biliary atresia. Pediatr. Surg. Int. 2017, 33, 1047–1052.
- Obayashi, J.; Tanaka, K.; Ohyama, K.; Manabe, S.; Nagae, H.; Shima, H.; Sato, H.; Furuta, S.; Wakisaka, M.; Koike, J.; et al. Relation between amount of bile ducts in portal canal and outcomes in biliary atresia. Pediatr. Surg. Int. 2016, 32, 833–838.

