Despite the fact that the etiology is far from being known, pediatric surgeries conducted last century worldwide were incredibly supportive for the clarification of the nosology of this disease [2,3,4]. Mainly, Morio Kasai, a Japanese pediatric surgeon, may be considered the pioneer of a specific pediatric surgery that may be offered to parents of a child with biliary atresia. His name is indelibly bound to the palliative surgical procedure he ideated in Japan [5]. The porto-enterostomy is indeed the major palliative procedure, which may be implemented before a liver is available for transplantation (orthotopic transplant or transplant of a liver from a recently deceased donor, a living donor transplant, or a split type of liver transplant) can be put forward. The biliary atresia is a very complex disease, not just complicated, and several genes have been investigated thoroughly. Some of these genes may be highly relevant to this disease, although their role is mostly not known.
2. Pathological Anatomy of the Biliary Atresia
The histology of the liver with biliary atresia is variable and depends on the stage when the biopsy is done substantially. At the early stage, fibrosis and biliary ductular proliferation, two main essential criteria, are present but may be very focal and missed at first glance. A subsequent liver biopsy may harbor more conclusive findings. However, the liver biopsy is essential to indicate if the main inflammatory component is at the level of the portal tracts or at acinar level, which may indicate neonatal hepatitis [
7,
14]. Thus, in biliary atresia, there are prominent histologic features, including a limited involvement of the liver acinus with very few multinucleated giant hepatocytes, which should have six or more nuclei. The limitation of acinus involvement is critical. At the late stage, fibrosis becomes more prominent, and the chances to be successful with a hepatic portoenterostomy are limited [
6,
26]. At the late stage, liver cirrhosis is usually encountered with pseudolobules and lobules surrounded by expanded portal tracts with biliary ductular proliferation (
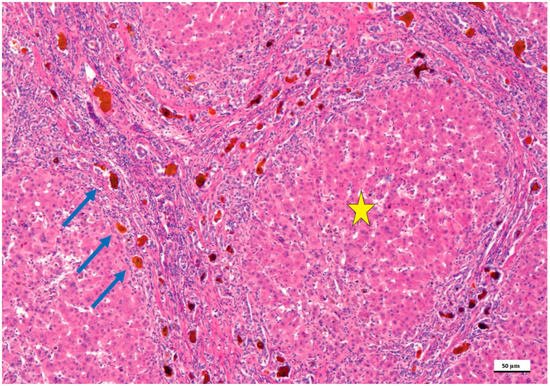
Figure 1).
Figure 1. Biliary atresia in a child treated with the Kasai portoenterostomy, which failed. In this microphotograph, the formation of lobules and pseudo-lobules (yellow star) is apparent indicating stage IV fibrosis/cirrhosis. The portal tracts are fused highlighting the porto-portal and centro-portal bridges. In these bridges (blue arrows), the intrahepatic biliary tract shows biliary ductal proliferations and lumens filled with bile, which appears brown (hematoxylin and eosin staining, 50× as original magnification, bar: 50 μm).
One of the most detailed studies on biliary atresia was performed at the Division of Surgery, Children’s Research Hospital, Kyoto Prefectural University of Medicine, Kyoto, Japan. In this clinic-pathological investigation, 31 patients with uncorrectable obstructive cholangiopathy underwent hepatic portoenterostomy and steroid therapy between 1988 and 2005 [
16]. An immunohistochemical investigation was performed using an antibody against keratin 19 of the intermediate filament family of the cytoskeleton. In this study, ductal plate malformation (DPM) typing was performed according to generally accepted criteria [
27]. Only type II DPM was considered relevant, and specimens were deemed as DPM-positive if a concentric cellular arrangement was detected. Shimadera et al. found that the presence of DPM in the liver of patients with biliary atresia predicts poor bile flow after hepatoportoenterostomy [
16]. This study was retrospective and included comparisons of preoperative characteristics, the postoperative jaundice period, and cumulative steroid doses between patients with and without DPM. Despite numerous reports on the DPM of biliary atresia, the influence of biliary remnants remains controversially discussed in the literature. During the re-examination of the microphotographic material, researchers identified abnormalities, such as cytomegalovirus expression in the placenta (
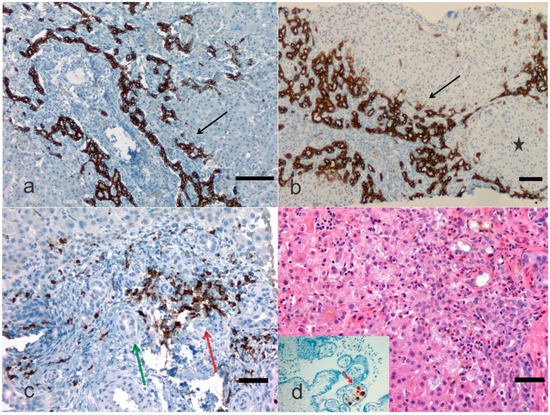
Figure 2). Visualization of cytokeratins (keratins) and neural cell adhesion molecules in the immature ductal cells by means of the immunohistochemical method can be a useful tool for the microscopic examination of the immature biliary structures in the liver. Russo et al. [
28] assessed the relative value of histologic features in 227 liver needle biopsies in discriminating between biliary atresia and other cholestatic disorders in infants enrolled in a prospective Childhood Liver Disease Research and Education Network (ChiLDReN) cohort study. These authors correlated histology with clinical findings in infants with and without biliary atresia. Also, Russo et al. reviewed 316 liver biopsies from clinically proven biliary atresia patients and correlated histologic features with total serum bilirubin 6 months post-Kasai or hepatoportoenterostomy and transplant-free survival up to 6 years. Logistic regression analysis determined that bile plugs in portal bile ducts/ductules, moderate to marked ductular reaction and portal stromal edema had the largest odds ratio for predicting biliary atresia vs. non- biliary atresia. Remarkably, the diagnostic accuracy of the needle liver biopsy was assessed to be 90.1%, whereas sensitivity and specificity for a diagnosis of biliary atresia were 88.4% and 92.7%, respectively. Russo et al. found that higher stages of fibrosis, a ductal plate configuration, moderate to marked bile duct injury, an older age at hepatoportoenterostomy and an elevated INR (international normalized ratio), which is a type of calculation based on prothrombin test results, were independently associated with a higher risk of transplantation [
28].
Figure 2. Biliary atresia with different clinical background. In this microphotograph panel, the bile ductular proliferations (arrows) are highlighted with a monoclonal antibody against cytokeratin or keratin 19 (a) and against cytokeratin or keratin 7 (b) by immunohistochemistry. Keratin-19 or cytokeratin-19 is a 40 kDa type I cytoskeletal protein that in humans is encoded by the KRT19 gene. In (b) the bile ductular proliferation is particularly pronounced (arrow) and a pseudonodule is noted (black star). In (c), an antibody against myeloperoxidase is carried out in an early case of biliary atresia highlighting the neutrophilic inflammation (red arrow) close to bile ducts (green arrow). Staining is either by immunohistochemistry (this case) or enzyme cytochemistry. Myeloperoxidase is the most sensitive and specific stain for myeloid leukemias and granulocytic sarcoma, but it is also very useful to stain neutrophils, particularly when their identification may be difficult to catch when other cells confuse the histology. In (d) a child with biliary atresia and positive cytomegalovirus (CMV) serology, but negative CMV immunohistochemistry, may disclose CMV positive cells in the placenta ((a), Bar 50 μm, 100× original magnification; (b), Bar 50 μm, 50× original magnification; (c), Bar 10 μm, 200× original magnification; (d), Bar 10 μm, 200× original magnification, and inset, 200× original magnification).
Apart from biliary atresia major causes of cholestatic liver disease in infants include choledochal cyst, Caroli’s syndrome, Alagille syndrome, and neonatal infection (Cytomegalovirus Herpesvirus Hepatotropic viruses, Human immunodeficiency virus, Parvovirus B19, Paramyxovirus, Enteric viruses, Rubella, Bacterial sepsis, Listeriosis, Toxoplasmosis, Syphilis). Also, disorders of carbohydrate metabolism, disorders of amino acid metabolism, disorders of glycolipid and lipid metabolism, disorders of glycoprotein metabolism, metal storage disorders, peroxisomal disorders, mitochondrial cytopathies, hereditary disorders of bilirubin metabolism, hereditary disorders of bile formation, disorders of bile acid biosynthesis, disorders of protein biosynthesis and targeting (e.g., α1-Antitrypsin deficiency) and miscellaneous disorders (e.g., Aagenaes syndrome, citrullinemia, type II X-linked adrenoleukodystrophy, shock/hypoperfusion, parenteral nutrition, fetal alcohol syndrome, drugs, Budd-Chiari syndrome, multiple hemangiomas) as well as neoplastic diseases (e.g., neonatal leukemia, neuroblastoma, hepatoblastoma, Langerhans cell histiocytosis, and erythrophagocytic lymphohistiocytosis). The broad differential diagnosis of jaundice in infants and children presupposes a good clinical and pathological correlation and periodic liver rounds. Since the evaluation of pediatric liver biopsies is quite distinct from that of adults, the liver pathologist needs to possess a good knowledge of biochemistry and metabolic disorders as well as dysmorphology understanding. The performance of all potentially necessary tests when a liver biopsy is carried out, prior arrangements should be in place between the clinician, the radiologist, and the laboratory to ensure adequate specimen processing and rapid workout. The laboratory should have liquid nitrogen on site or, better, at the bedside. Cores of tissue, up to 2 cm in length, should be snap frozen in liquid nitrogen refrigerated isopentane immediately to preserve tissue integrity and mRNA (messenger ribonucleic acid). The tissue fragments should then be placed within a specimen vial and a small portion of tissue may be placed in glutaraldehyde for electron microscopy processing, and a portion can be sent to the biochemistry laboratory for specific requests.