 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jian-Ji Zhong | + 4133 word(s) | 4133 | 2022-01-25 03:30:30 | | | |
| 2 | Vivi Li | -4 word(s) | 4129 | 2022-02-11 02:28:44 | | |
Video Upload Options
Visible-light photoredox catalysis has been established as a popular and powerful tool for organic transformations owing to its inherent characterization of environmental friendliness and sustainability in the past decades. The thiol-ene/yne reactions, the direct hydrothiolation of alkenes/alkynes with thiols, represents one of the most efficient and atom-economic approaches for the carbon-sulfur bonds construction. In traditional methodologies, harsh conditions such as stoichiometric reagents or a specialized UV photo-apparatus were necessary suffering from various disadvantages. In particular, visible-light photoredox catalysis has also been demonstrated to be a greener and milder protocol for the thiol-ene/yne reactions in recent years. Additionally, unprecedented advancements have been achieved in this area during the past decade.
1. Introduction
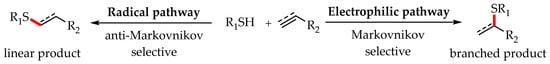
2. Photoredox Catalytic Anti-Markovnikov-Selective Thiol-Ene/Yne Reactions
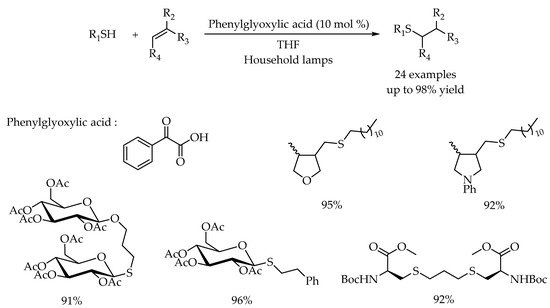
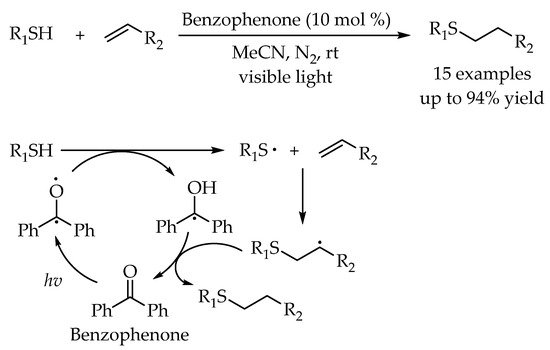
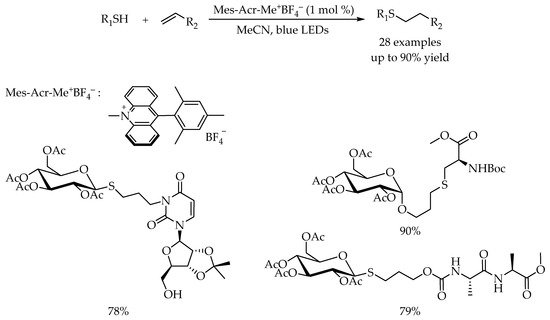
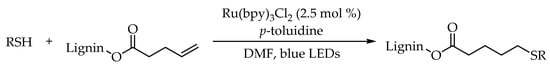
2.1. Photocatalytic Anti-Markovnikov Thiol-Ene Reactions for Synthesis of Alkyl Thioethers
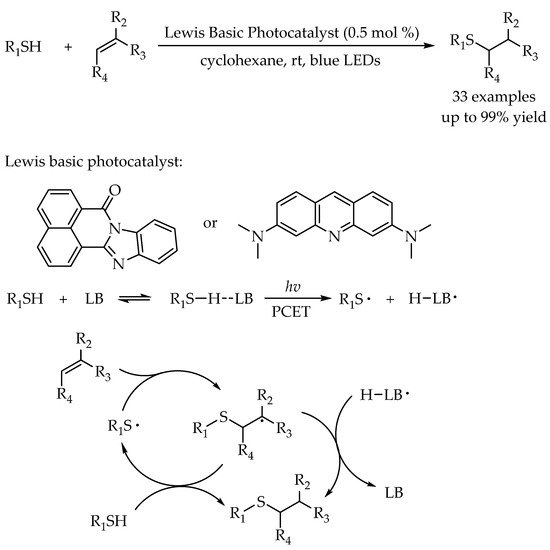
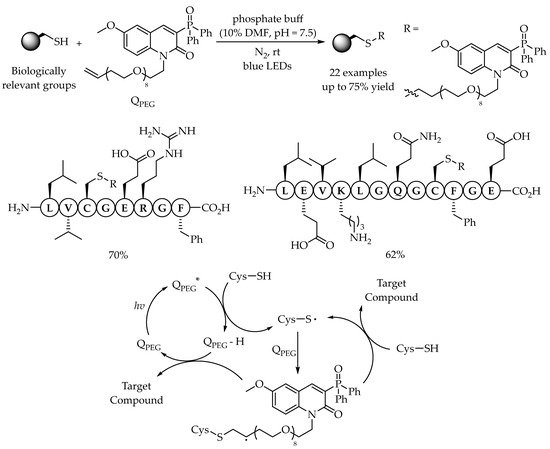
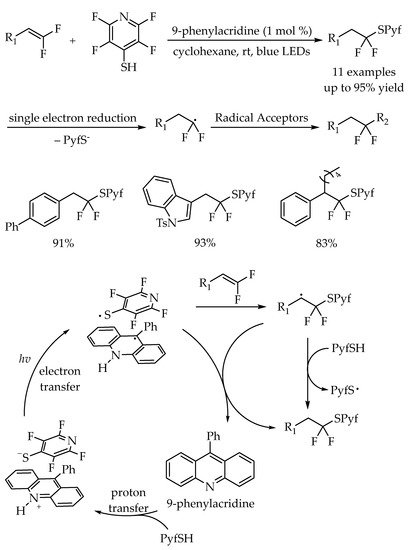
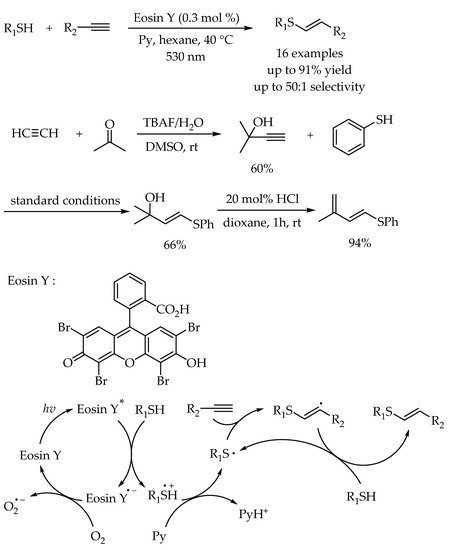
2.2. Photocatalytic Anti-Markovnikov Thiol-Yne Reactions for Synthesis of Alkenyl Thioethers
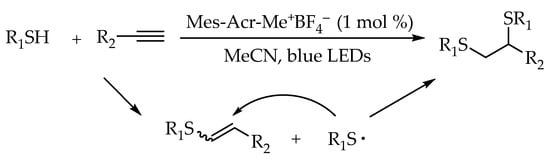
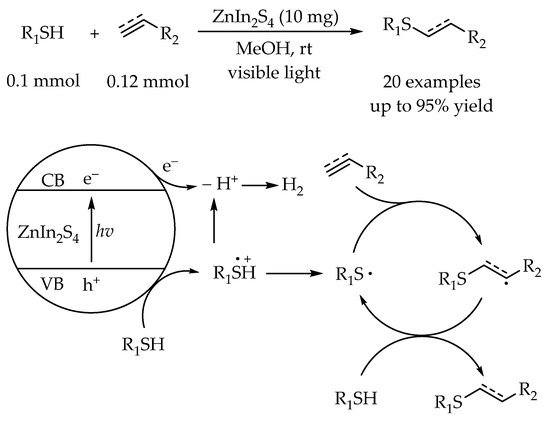
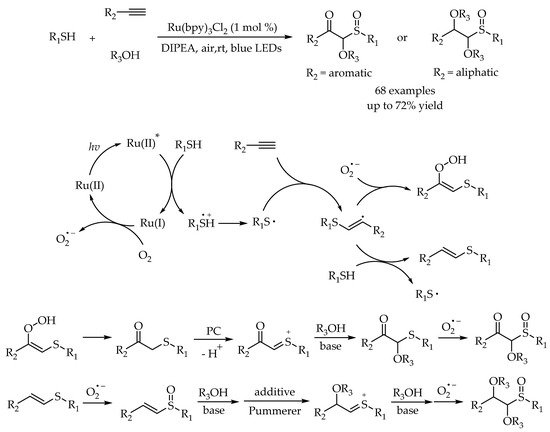
2.3. Photocatalytic Anti-Markovnikov Thiol-Ene/Yne Reactions for Synthesis of β-Hydroxysulfides/β-Keto Sulfides
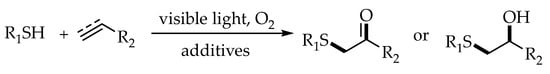
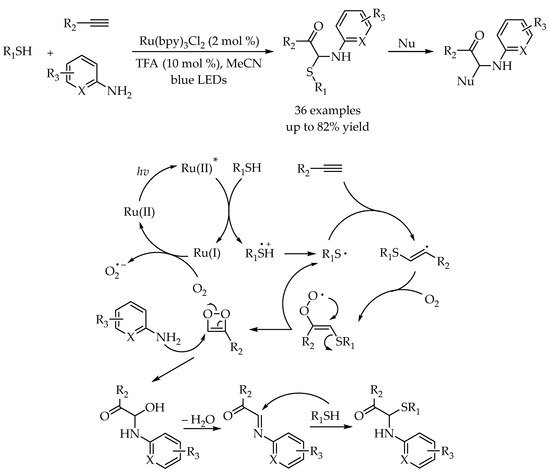
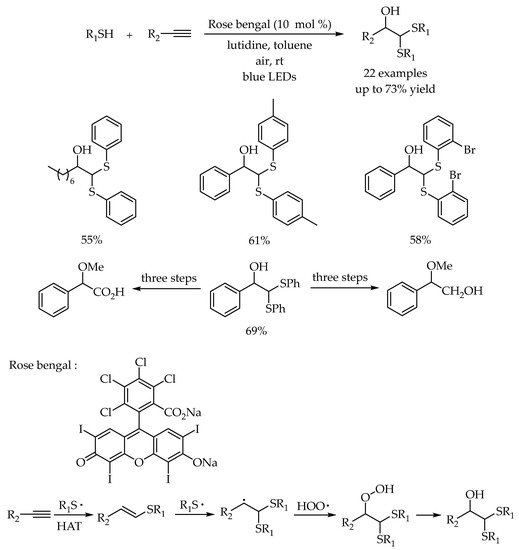
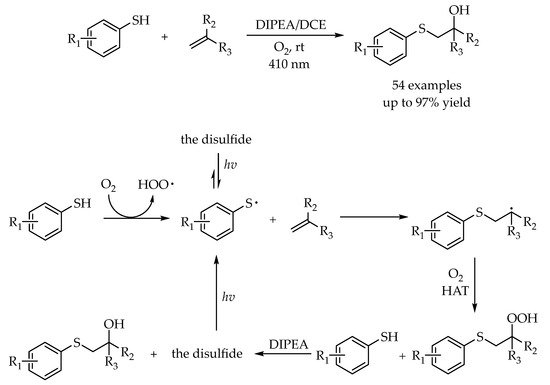
2.4. Photocatalytic Anti-Markovnikov Thiol–Ene/Yne Reactions for Synthesis of Sulfoxides
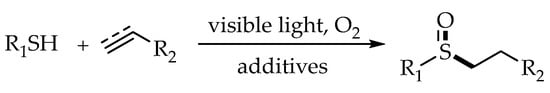
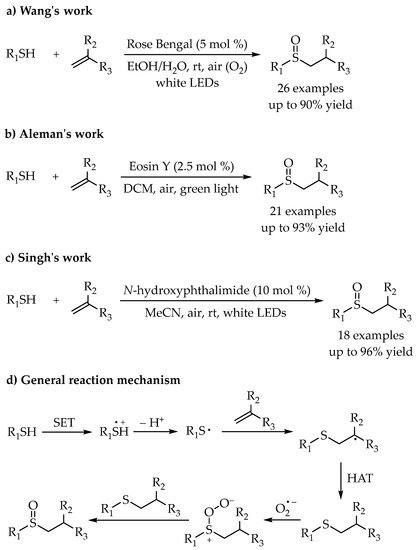
3. Photoredox Catalytic Markovnikov-Selective Thiol-Ene/Yne Reactions
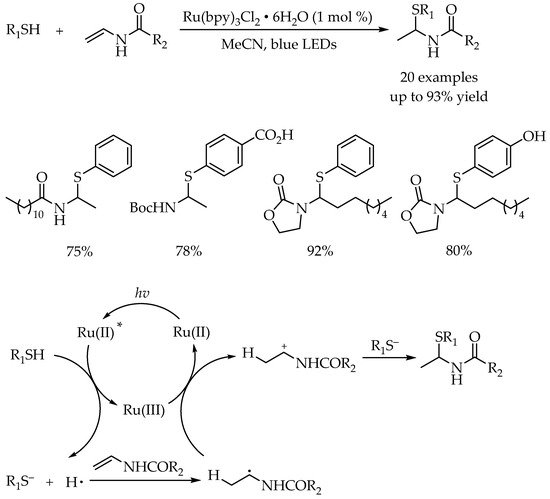
3.1. Photocatalytic Markovnikov Thiol-Ene Reactions for Synthesis of Branched Alkyl Thioethers
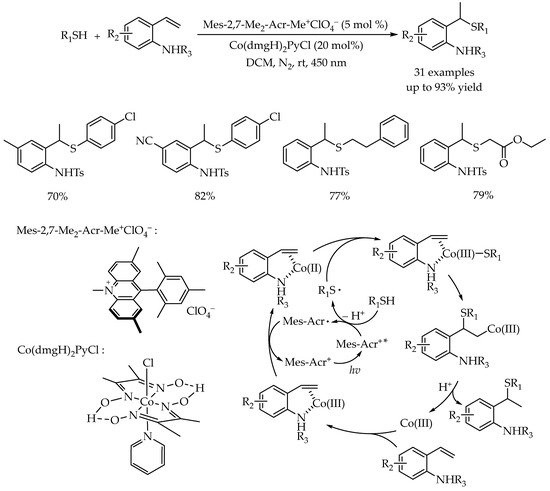
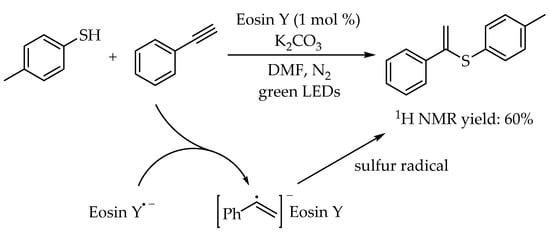
3.2. Photocatalytic Markovnikov Thiol-Yne Reactions for Synthesis of Branched Alkenyl Thioethers
References
- Jacob, C. A Scent of Therapy: Pharmacological Implications of Natural Products Containing Redox-Active Sulfur Atoms. Nat. Prod. Rep. 2006, 23, 851–863.
- Clayden, J.; MacLellan, P. Asymmetric Synthesis of Tertiary Thiols and Thioethers. Beilstein J. Org. Chem. 2011, 7, 582–595.
- Feng, M.; Tang, B.; Liang, S.H.; Jiang, X. Sulfur Containing Scaffolds in Drugs: Synthesis and Application in Medicinal Chemistry. Curr. Top. Med. Chem. 2016, 16, 1200–1216.
- Wang, N.; Saidhareddy, P.; Jiang, X. Construction of Sulfur-Containing Moieties in the Total Synthesis of Natural Products. Nat. Prod. Rep. 2020, 37, 246–275.
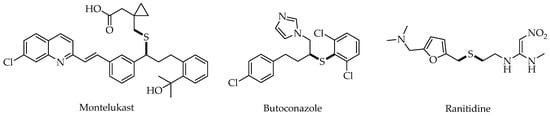
- Han, J.; Jia, Y.; Takeda, K.; Shiraishi, Y.; Okamoto, M.; Dakhama, A.; Gelfand, E.W. Montelukast during Primary Infection Prevents Airway Hyperresponsiveness and Inflammation after Reinfection with Respiratory Syncytial Virus. Am. J. Respir. Crit. Care Med. 2010, 182, 455–463.
- Barré, J.; Sabatier, J.-M.; Annweiler, C. Montelukast Drug May Improve COVID-19 Prognosis: A Review of Evidence. Front. Pharmacol. 2020, 11, 1344.
- Qin, F.; Wang, Q.; Zhang, C.; Fang, C.; Zhang, L.; Chen, H.; Zhang, M.; Cheng, F. Efficacy of Antifungal Drugs in the Treatment of Vulvovaginal Candidiasis: A Bayesian Network Meta-Analysis. Infect. Drug Resist. 2018, 11, 1893–1901.
- Hacioglu, M.; Guzel, C.B.; Savage, P.B.; Tan, A.S.B. Antifungal Susceptibilities, in Vitro Production of Virulence Factors and Activities of Ceragenins against Candida Spp. Isolated from Vulvovaginal Candidiasis. Med. Mycol. 2019, 57, 291–299.
- Johansson, S.; Read, J.; Oliver, S.; Steinberg, M.; Li, Y.; Lisbon, E.; Mathews, D.; Leese, P.T.; Martin, P. Pharmacokinetic Evaluations of the Co-Administrations of Vandetanib and Metformin, Digoxin, Midazolam, Omeprazole or Ranitidine. Clin. Pharmacokinet. 2014, 53, 837–847.
- McGarrigle, E.M.; Myers, E.L.; Illa, O.; Shaw, M.A.; Riches, S.L.; Aggarwal, V.K. Chalcogenides as Organocatalysts. Chem. Rev. 2007, 107, 5841–5883.
- Hoyle, C.E.; Lowe, A.B.; Bowman, C.N. Thiol-Click Chemistry: A Multifaceted Toolbox for Small Molecule and Polymer Synthesis. Chem. Soc. Rev. 2010, 39, 1355–1387.
- Arrayás, R.G.; Carretero, J.C. Chiral Thioether-Based Catalysts in Asymmetric Synthesis: Recent Advances. Chem. Commun. 2011, 47, 2207–2211.
- Yuan, D.; Huynh, H.V. Sulfur-Functionalized N-Heterocyclic Carbene Complexes of Pd(II): Syntheses, Structures and Catalytic Activities. Molecules 2012, 17, 2491–2517.
- Lynch, D.M.; Scanlan, E.M. Thiyl Radicals: Versatile Reactive Intermediates for Cyclization of Unsaturated Substrates. Molecules 2020, 25, 3094.
- Gress, A.; Völkel, A.; Schlaad, H. Thio-Click Modification of Poly. Macromolecules 2007, 40, 7928–7933.
- ten Brummelhuis, N.; Diehl, C.; Schlaad, H. Thiol−Ene Modification of 1,2-Polybutadiene Using UV Light or Sunlight. Macromolecules 2008, 41, 9946–9947.
- Hoyle, C.E.; Bowman, C.N. Thiol-Ene Click Chemistry. Angew. Chem. Int. Ed. 2010, 49, 1540–1573.
- Scanlan, E.; Corcé, V.; Malone, A. Synthetic Applications of Intramolecular Thiol-Ene “Click” Reactions. Molecules 2014, 19, 19137–19151.
- Sánchez-Fernández, E.M.; García-Moreno, M.I.; García-Hernández, R.; Padrón, J.M.; García Fernández, J.M.; Gamarro, F.; Ortiz Mellet, C. Thiol-Ene “Click” Synthesis and Pharmacological Evaluation of C-Glycoside Sp2-Iminosugar Glycolipids. Molecules 2019, 24, 2882.
- Hearon, K.; Nash, L.D.; Rodriguez, J.N.; Lonnecker, A.T.; Raymond, J.E.; Wilson, T.S.; Wooley, K.L.; Maitland, D.J. A High-Performance Recycling Solution for Polystyrene Achieved by the Synthesis of Renewable Poly(Thioether) Networks Derived from D-Limonene. Adv. Mater. 2014, 26, 1552–1558.
- Kumar, R.; Saima; Shard, A.; Andhare, N.H.; Richa; Sinha, A.K. Thiol-Ene “Click” Reaction Triggered by Neutral Ionic Liquid: The “Ambiphilic” Character of Br in the Regioselective Nucleophilic Hydrothiolation. Angew. Chem. Int. Ed. 2015, 54, 828–832.
- Healy, J.; Rasmussen, T.; Miller, S.; Booth, I.R.; Conway, S.J. The Photochemical Thiol–Ene Reaction as a Versatile Method for the Synthesis of Glutathione S-Conjugates Targeting the Bacterial Potassium Efflux System Kef. Org. Chem. Front. 2016, 3, 439–446.
- Vanslambrouck, S.; Riva, R.; Ucakar, B.; Préat, V.; Gagliardi, M.; Molin, D.G.M.; Lecomte, P.; Jérôme, C. Thiol-Ene Reaction: An Efficient Tool to Design Lipophilic Polyphosphoesters for Drug Delivery Systems. Molecules 2021, 26, 1750.
- Shi, L.; Xia, W. Photoredox Functionalization of C–H Bonds Adjacent to a Nitrogen Atom. Chem. Soc. Rev. 2012, 41, 7687–7697.
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363.
- Ghosh, I.; Marzo, L.; Das, A.; Shaikh, R.; König, B. Visible Light Mediated Photoredox Catalytic Arylation Reactions. Acc. Chem. Res. 2016, 49, 1566–1577.
- Kärkäs, M.D.; Porco, J.A.; Stephenson, C.R.J. Photochemical Approaches to Complex Chemotypes: Applications in Natural Product Synthesis. Chem. Rev. 2016, 116, 9683–9747.
- Skubi, K.L.; Blum, T.R.; Yoon, T.P. Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev. 2016, 116, 10035–10074.
- Chen, B.; Wu, L.-Z.; Tung, C.-H. Photocatalytic Activation of Less Reactive Bonds and Their Functionalization via Hydrogen-Evolution Cross-Couplings. Acc. Chem. Res. 2018, 51, 2512–2523.
- Zhong, J.-J.; To, W.-P.; Liu, Y.; Lu, W.; Che, C.-M. Efficient Acceptorless Photo-Dehydrogenation of Alcohols and N-Heterocycles with Binuclear Platinum(II) Diphosphite Complexes. Chem. Sci. 2019, 10, 4883–4889.
- Qi, X.-K.; Zhang, H.; Pan, Z.-T.; Liang, R.-B.; Zhu, C.-M.; Li, J.-H.; Tong, Q.-X.; Gao, X.-W.; Wu, L.-Z.; Zhong, J.-J. Photoinduced Synthesis of Fluorinated DibenzAzepines via Radical Triggered Cyclization. Chem. Commun. 2019, 55, 10848–10851.
- Zhang, H.; Xiao, Q.; Qi, X.-K.; Gao, X.-W.; Tong, Q.-X.; Zhong, J.-J. Selective Photoredox Decarboxylation of α-Ketoacids to Allylic Ketones and 1,4-Dicarbonyl Compounds Dependent on Cobaloxime Catalysis. Chem. Commun. 2020, 56, 12530–12533.
- Xu, H.; Zhang, H.; Tong, Q.-X.; Zhong, J.-J. Photoredox/Cobaloxime Co-Catalyzed Allylation of Amines and Sulfonyl Hydrazines with Olefins to Access α-Allylic Amines and Allylic Sulfones. Org. Biomol. Chem. 2021, 19, 8227–8231.
- Lu, F.-D.; Chen, J.; Jiang, X.; Chen, J.-R.; Lu, L.-Q.; Xiao, W.-J. Recent Advances in Transition-Metal-Catalysed Asymmetric Coupling Reactions with Light Intervention. Chem. Soc. Rev. 2021, 50, 12808–12827.
- Xiao, Q.; Lu, M.; Deng, Y.; Jian, J.-X.; Tong, Q.-X.; Zhong, J.-J. Photoinduced Radical Cascade Cyclization: A Metal-Free Approach to Access Difluoroalkylated Dioxodibenzothiazepines. Org. Lett. 2021, 23, 9303–9308.
- Tyson, E.L.; Ament, M.S.; Yoon, T.P. Transition Metal Photoredox Catalysis of Radical Thiol-Ene Reactions. J. Org. Chem. 2013, 78, 2046–2050.
- Keylor, M.H.; Park, J.E.; Wallentin, C.-J.; Stephenson, C.R.J. Photocatalytic Initiation of Thiol–Ene Reactions: Synthesis of Thiomorpholin-3-Ones. Tetrahedron 2014, 70, 4264–4269.
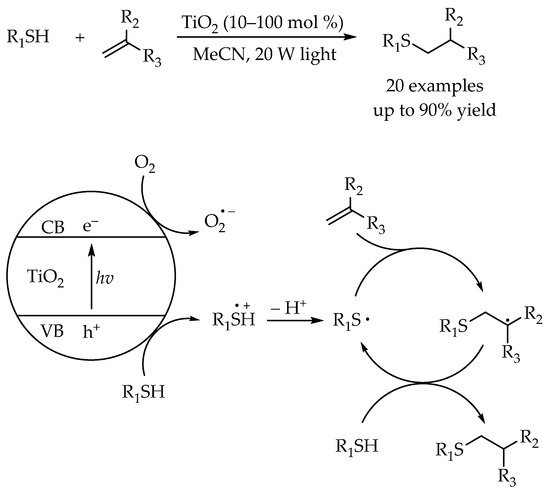
- Bhat, V.T.; Duspara, P.A.; Seo, S.; Abu Bakar, N.S.B.; Greaney, M.F. Visible Light Promoted Thiol-Ene Reactions Using Titanium Dioxide. Chem. Commun. 2015, 51, 4383–4385.
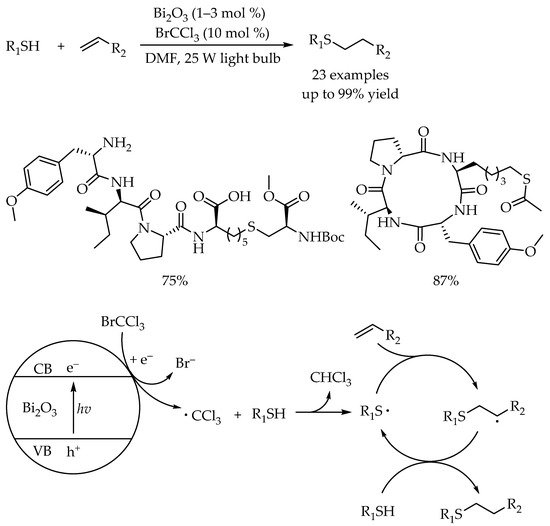
- Fadeyi, O.O.; Mousseau, J.J.; Feng, Y.; Allais, C.; Nuhant, P.; Chen, M.Z.; Pierce, B.; Robinson, R. Visible-Light-Driven Photocatalytic Initiation of Radical Thiol–Ene Reactions Using Bismuth Oxide. Org. Lett. 2015, 17, 5756–5759.
- Limnios, D.; Kokotos, C.G. Photoinitiated Thiol-Ene “Click” Reaction: An Organocatalytic Alternative. Adv. Synth. Catal. 2017, 359, 323–328.
- Singh, M.; Yadav, A.K.; Yadav, L.D.S.; Singh, R.K.P. Visible Light Photocatalysis with Benzophenone for Radical Thiol-Ene Reactions. Tetrahedron Lett. 2017, 58, 2206–2208.
- Zhao, G.; Kaur, S.; Wang, T. Visible-Light-Mediated Thiol–Ene Reactions through Organic Photoredox Catalysis. Org. Lett. 2017, 19, 3291–3294.
- Liu, H.; Chung, H. Visible-Light Induced Thiol–Ene Reaction on Natural Lignin. ACS Sustain. Chem. Eng. 2017, 5, 9160–9168.
- Levin, V.V.; Dilman, A.D. Visible-Light-Mediated Organocatalyzed Thiol–Ene Reaction Initiated by a Proton-Coupled Electron Transfer. J. Org. Chem. 2019, 84, 8337–8343.
- Choi, H.; Kim, M.; Jang, J.; Hong, S. Visible-Light-Induced Cysteine-Specific Bioconjugation: Biocompatible Thiol–Ene Click Chemistry. Angew. Chem. Int. Ed. 2020, 59, 22514–22522.
- Zubkov, M.O.; Kosobokov, M.D.; Levin, V.V.; Kokorekin, V.A.; Korlyukov, A.A.; Hu, J.; Dilman, A.D. A Novel Photoredox-Active Group for the Generation of Fluorinated Radicals from Difluorostyrenes. Chem. Sci. 2020, 11, 737–741.
- Zalesskiy, S.S.; Shlapakov, N.S.; Ananikov, V.P. Visible Light Mediated Metal-Free Thiol–Yne Click Reaction. Chem. Sci. 2016, 7, 6740–6745.
- Kaur, S.; Zhao, G.; Busch, E.; Wang, T. Metal-Free Photocatalytic Thiol–Ene/Thiol–Yne Reactions. Org. Biomol. Chem. 2019, 17, 1955–1961.
- Li, Y.; Cai, J.; Hao, M.; Li, Z. Visible Light Initiated Hydrothiolation of Alkenes and Alkynes over ZnIn2S4. Green Chem. 2019, 21, 2345–2351.
- Chalotra, N.; Rizvi, M.A.; Shah, B.A. Photoredox-Mediated Generation of Gem -Difunctionalized Ketones: Synthesis of α,α-Aminothioketones. Org. Lett. 2019, 21, 4793–4797.
- Manzer Manhas, F.; Kumar, J.; Raheem, S.; Thakur, P.; Rizvi, M.A.; Shah, B.A. Photoredox-Mediated Synthesis of β-Hydroxydithioacetals from Terminal Alkynes. ChemPhotoChem 2021, 5, 235–239.
- Shi, J.; Gao, X.-W.; Tong, Q.-X.; Zhong, J.-J. Light-Promoted and Tertiary-Amine-Assisted Hydroxysulfenylation of Alkenes: Selective and Direct One-Pot Synthesis of β-Hydroxysulfides. J. Org. Chem. 2021, 86, 12922–12931.
- Cui, H.; Wei, W.; Yang, D.; Zhang, Y.; Zhao, H.; Wang, L.; Wang, H. Visible-Light-Induced Selective Synthesis of Sulfoxides from Alkenes and Thiols Using Air as the Oxidant. Green Chem. 2017, 19, 3520–3524.
- Guerrero-Corella, A.; María Martinez-Gualda, A.; Ahmadi, F.; Ming, E.; Fraile, A.; Alemán, J. Thiol–Ene/Oxidation Tandem Reaction under Visible Light Photocatalysis: Synthesis of Alkyl Sulfoxides. Chem. Commun. 2017, 53, 10463–10466.
- Singh, M.; Yadav, A.K.; Yadav, L.D.S.; Singh, R.K.P. Visible-Light-Activated Selective Synthesis of Sulfoxides via Thiol-Ene/Oxidation Reaction Cascade. Tetrahedron Lett. 2018, 59, 450–453.
- Kumar, J.; Ahmad, A.; Rizvi, M.A.; Ganie, M.A.; Khajuria, C.; Shah, B.A. Photoredox-Mediated Synthesis of Functionalized Sulfoxides from Terminal Alkynes. Org. Lett. 2020, 22, 5661–5665.
- Gossage, R.A.; van de Kuil, L.A.; van Koten, G. Diaminoarylnickel(II) “Pincer” Complexes: Mechanistic Considerations in the Kharasch Addition Reaction, Controlled Polymerization, and Dendrimeric Transition Metal Catalysts. Acc. Chem. Res. 1998, 31, 423–431.
- Wille, U. Radical Cascades Initiated by Intermolecular Radical Addition to Alkynes and Related Triple Bond Systems. Chem. Rev. 2013, 113, 813–853.
- Barman, E.; Hourizadeh, J.; Lim, D. Visible Light Photoredox-Catalyzed Hydrothiolation of Enamides and Enecarbamates. Tetrahedron Lett. 2020, 61, 152201.
- Xiao, Q.; Zhang, H.; Li, J.-H.; Jian, J.-X.; Tong, Q.-X.; Zhong, J.-J. Directing-Group-Assisted Markovnikov-Selective Hydrothiolation of Styrenes with Thiols by Photoredox/Cobalt Catalysis. Org. Lett. 2021, 23, 3604–3609.
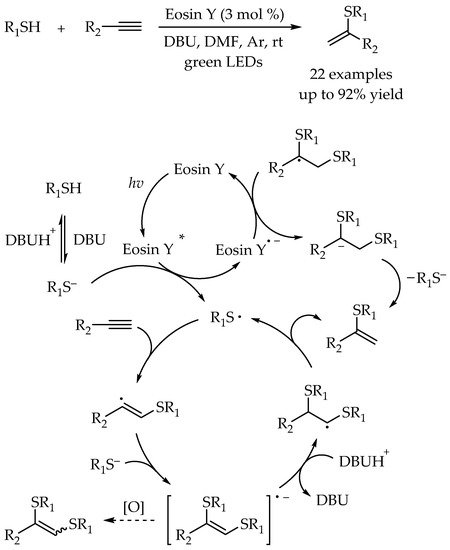
- Wang, H.; Lu, Q.; Chiang, C.-W.; Luo, Y.; Zhou, J.; Wang, G.; Lei, A. Markovnikov-Selective Radical Addition of S-Nucleophiles to Terminal Alkynes through a Photoredox Process. Angew. Chem. Int. Ed. 2017, 56, 595–599.
- Burykina, J.V.; Shlapakov, N.S.; Gordeev, E.G.; König, B.; Ananikov, V.P. Selectivity Control in Thiol–Yne Click Reactions via Visible Light Induced Associative Electron Upconversion. Chem. Sci. 2020, 11, 10061–10070.

