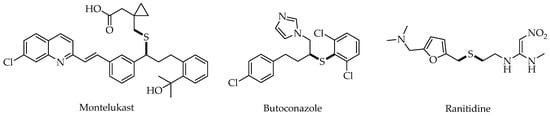
In most cases, due to the high reactivity of sulfur, the thiol-ene/yne reactions tend to proceed via a radical pathway to provide the anti-Markovnikov products by visible-light photoredox catalysis. Since Yoon and Stephenson’s pioneering works, various photocatalytic systems with anti-Markovnikov selectivity have been reported for synthesis of the diversity of organosulfur compounds.
2.1. Photocatalytic Anti-Markovnikov Thiol-Ene Reactions for Synthesis of Alkyl Thioethers
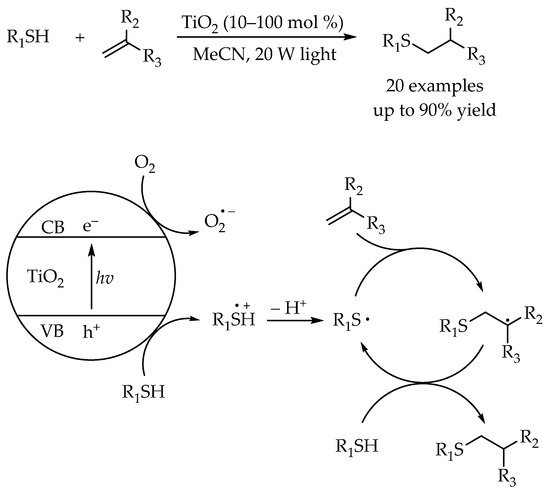
In 2015, Greaney’s group developed a visible-light-promoted thiol-ene reaction for synthesis of alkyl thioethers using TiO
2 as photocatalyst [
38]. Compared with the commonly used transition-metal-based photocatalysts, herein, TiO
2 was cheap, readily accessible, and provided the advantage of catalyst recyclability. This work proved that besides the transition-metal complexes, inorganic semiconductors could also act as an efficient photocatalyst in the thiol-ene reaction. According to the proposed mechanism, the reaction was initiated by a photoinduced electron transfer between the thiol and the excited TiO
2 to generate a thiyl radical. Subsequently, the anti-Markovnikov-selective addition of the thiyl radical to the alkene would provide an alkyl radical which could abstract hydrogen atom from an unreacted thiol to generate the corresponding alkyl sulfides (
Scheme 2).
Scheme 2. Visible-light-promoted thiol-ene reaction using titanium dioxide.
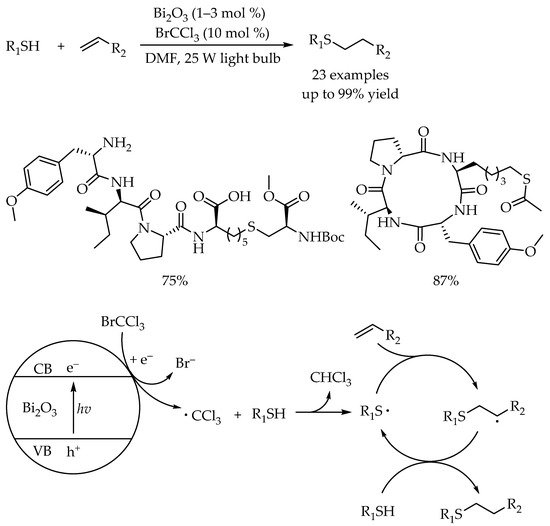
In the same year, Fadeyi’s group reported a visible-light photocatalytic anti-Markovnikov hydrothiolation reaction for alkyl sulfides synthesis using another nontoxic and inexpensive inorganic semiconductor Bi
2O
3, with broad functional group compatibility [
39]. In contrast to Greaney’s work, a variety of complex biologically relevant scaffolds such as linear tetrapeptide or cyclic tetrapeptide could readily undergo late-stage functionalization in this protocol, demonstrating the practicability of the protocol. The plausible mechanism showed that the photoinduced single electron transfer (SET) between Bi
2O
3 and bromotrichloromethane (BrCCl
3) would result in the formation of trichloromethyl radicals, which acted as a radical mediator to abstract a hydrogen atom from the thiol to generate a thiyl radical for inducing the radical thiol-ene reaction to access the corresponding anti-Markovnikov products (
Scheme 3).
Scheme 3. Visible-light-photocatalyzed radical thiol-ene reaction using bismuth oxide.
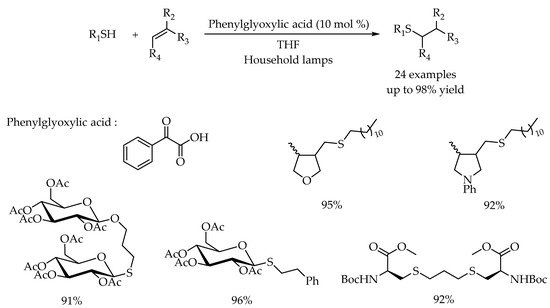
Transition-metal-based complexes were commonly employed as a catalyst in the photoredox catalyzed thiol-ene reactions. However, a problem that cannot be ignored is the intrinsic disadvantages of transition-metal complexes such as high cost, potential toxicity, and problematic removal of residual metal impurities. An alternative method was the introduction of inexpensive and less toxic metal-free organophotocatalyst. Therefore, Kokotos and co-workers reported a metal-free photoredox catalytic method for the radical thiol-ene reaction [
40]. Phenylglyoxylic acid was proved to be the most efficient organophotocatalyst providing the corresponding anti-Markovnikov alkyl thioethers in good to excellent yields. Besides the simple olefins, dienes could also serve as compatible partners. When diallyl ether and
N,
N-diallylaniline were employed as the substrates, a radical cascade cyclization happened to provide the alkyl sulfides with a tetrahydrofuran or pyrrolidine moiety in 95% and 92% yields, respectively. Further experiments demonstrated that such a mild and green protocol could be successfully applied to the modification of peptide and glycoside, and they also showed broad functional group tolerance (
Scheme 4).
Scheme 4. Visible-light-photocatalyzed thiol-ene reaction by phenylglyoxylic acid.
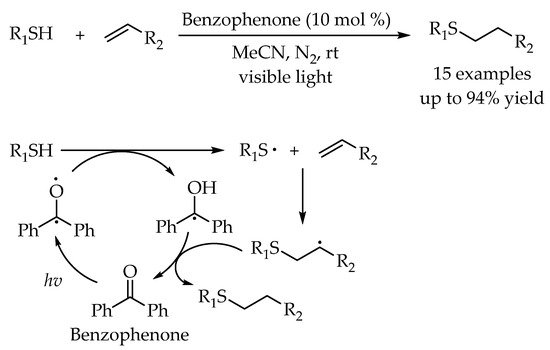
Furthermore, Singh and co-workers utilized benzophenone as an inexpensive organophotocatalyst to promote the carbon-sulfur bond formation through the reaction of thiols and olefins under visible-light irradiation [
41]. Diverse anti-Markovnikov adducts of thiols and olefins were obtained in good to excellent yields. For the proposed reaction pathway, the interaction between the thiol and the excited-state benzophenone could generate a thiyl radical and the ketyl radical by a hydrogen atom transfer (HAT) process. Then, the thiyl radical addition to the olefins furnished a new alkyl radical, which could abstract a hydrogen atom from the ketyl radical to fulfill the photocatalytic cycle and afford the alkyl sulfides (
Scheme 5).
Scheme 5. Visible-light-photocatalyzed thiol-ene reaction by benzophenone.
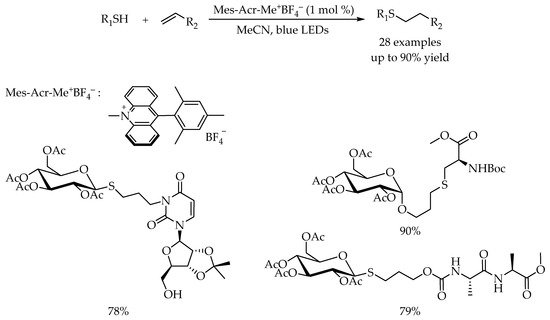
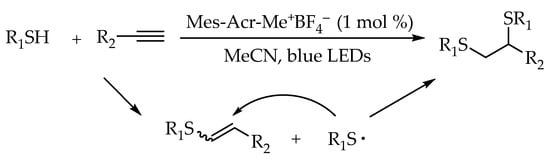
In 2017, Wang and co-workers reported a visible-light-promoted radical hydrothiolation of alkenes with thiols using 9-mesityl-10-methylacridinium tetrafluoroborate as photocatalyst [
42]. The reaction proceeded smoothly with a variety of alkenes and thiols, giving the alkyl thioethers in excellent yields. Moreover, examples of the synthesis of glycoconjugates between glycosyl thiols and amino acid derivatives demonstrated the potential applicability of this strategy. Particularly, the coupling of glucose thiol with a nucleoside derivatives and dipeptide derivatives could afford the glyconucleoside and glycopeptide in good yields (
Scheme 6).
Scheme 6. Visible-light-photocatalyzed thiol-ene reaction using 9-mesityl-10-methylacridinium tetrafluoroborate.
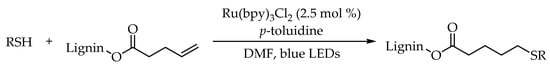
Polymeric modification of lignin was necessary. For this target, Chung’s group developed a lignin-involved visible-light-photocatalyzed thiol-ene reaction using Ru(bpy)
3Cl
2 as a photocatalyst [
43]. This low energy and environmentally friendly protocol was tolerant to a variety of functional groups, resulting in a high conversion of alkenes with reaction yields from 81% to 97%. Moreover, excellent reaction efficiency could also be achieved even after 4 h irradiation of natural sunlight (
Scheme 7). This protocol provided a simple and efficient way for constructing the structural complexity of lignin containing sulfur functional groups.
Scheme 7. Visible-light-photocatalyzed thiol-ene reaction for modification of natural lignin.
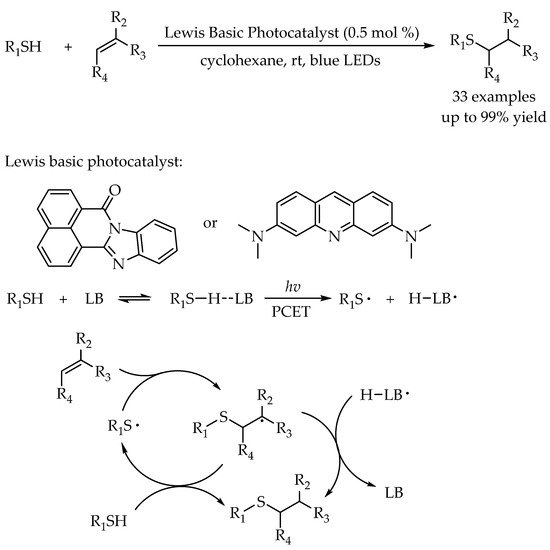
As mentioned above, in the radical thiol-ene reaction, a key species is the thiyl radical. In some cases, the sacrificial additive such as bromotrichloromethane has to be introduced to serve as a direct hydrogen atom transfer (HAT) reagent to activate the thiol for generating a thiyl radical. To avoid the use of sacrificial reagents, Dilman’s group utilized the photoactive Lewis basic species as a photocatalyst [
44]. A different reaction pathway, proton coupled electron transfer (PCET), was proposed. The interaction between the thiols and the excited Lewis basic catalyst under blue-light irradiation lead to the formation of thiyl radical for efficient radical thiol-ene reaction to furnish the anti-Markovnikov alkyl sulfides (
Scheme 8). This protocol exhibited a broad substrate scope and wide functional group compatibility. Terminal and internal alkenes, aliphatic and aromatic thiols could undergo smoothly in the reaction.
Scheme 8. Visible-light-photocatalyzed thiol-ene reaction via a proton-coupled electron transfer process.
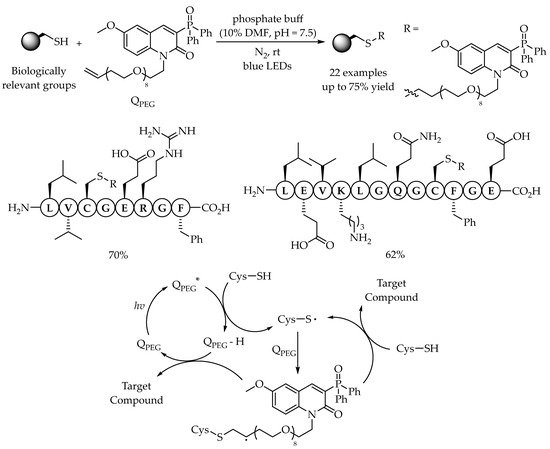
As one of the most promising cysteine modification methods, radical thiol-ene reaction for C-S bond formation was still limited because of the need for biocompatible reaction conditions. In 2020, Hong’s group discovered a visible-light-induced biocompatible thiol-ene reaction by using a fluorescent photosensitizer Q
PEG for cysteine-specific bioconjugation for the installation of Q
PEG [
45]. The exclusion of external oxidant and base in this catalytic system inhibited the undesired reaction of peptide and protein substrates with high efficiency and functional group tolerance. Importantly, the fragments of endogenous proteins derived from insulin and Src kinase could be successfully conjugated with Q
PEG with the yields of 70% and 62%, respectively. From the mechanistic studies, it was assumed that, under blue LEDs irradiation, the triplet excited state of Q
PEG could abstract a hydrogen atom from the cysteine forming a thiyl radical. After the addition of the thiyl radical to the alkene Q
PEG, an alkyl radical intermediate could be formed, which would abstract a hydrogen atom from another thiol to give rise to the corresponding alkyl sulfide. Alternatively, this alkyl radical could also abstract a hydrogen atom from the intermediate Q
PEG-H to form the product and regenerate the ground-state Q
PEG to close the catalytic cycle (
Scheme 9).
Scheme 9. Cysteine-specific bioconjugation by photocatalytic thiol-ene reaction under biocompatible conditions.
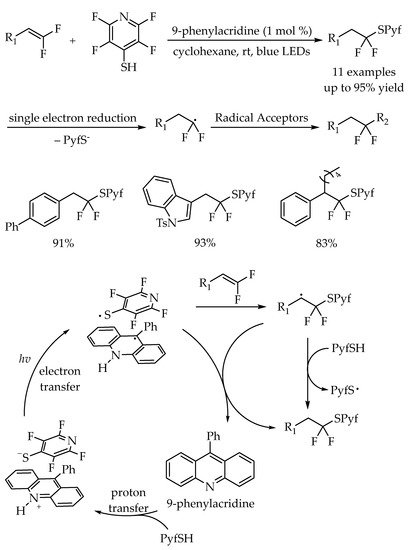
Recently, the thioethers bearing 4-tetrafluoropyridinylthio and difluoroalkyl groups have been used as precursors to form fluorinated carbon radicals for the construction of difluoroalkylated compounds by Dilman and co-workers [
46]. The synthesis of the thioethers involved the photocatalytic thiol-ene reaction using readily accessible difluorostyrenes with tetrafluoropyridine-4-thiol (PyfSH). This protocol was well tolerant of a series of difluoroalkenes bearing aromatic or heteroaromatic groups with good to excellent yields. In addition, a large scale reaction could also undergo smoothly in this protocol. Based on the studies, the authors believed that the generation of thioethers proceeded via a proton coupled electron transfer (PCET) mechanism. The combination of 9-phenylacridine and PyfSH (proton transfer, PT) providing a red colored salt, which could undergo a single electron transfer (SET) process to afford the thiophenol radical under visible light irradiation. After the radical addition to the double bond of difluorostyrenes, the resulting benzyl radical could abstract a hydrogen atom either from the N-H acridinium radical intermediate or from the starting material PyfSH. The measurement of the reduction potential of thioethers indicated that they could participate in a series of reactions with silyl enol ethers, alkenes, nitrones and an alkenyl trifluoroborate (
Scheme 10).
Scheme 10. Photocatalyzed thiol-ene reaction for difluorostyrenes functionalization with tetrafluoropyridine-4-thiol.
2.2. Photocatalytic Anti-Markovnikov Thiol-Yne Reactions for Synthesis of Alkenyl Thioethers
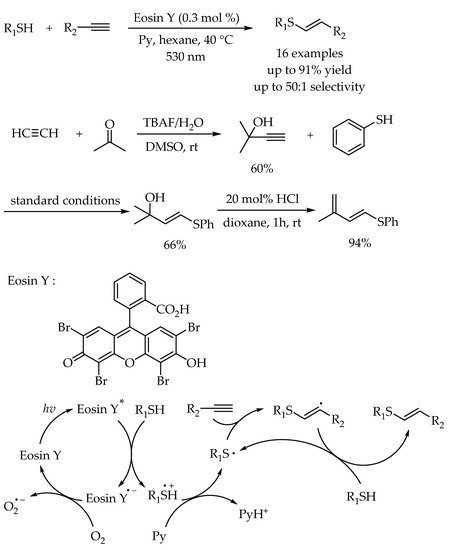
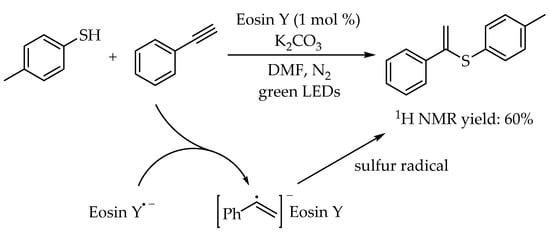
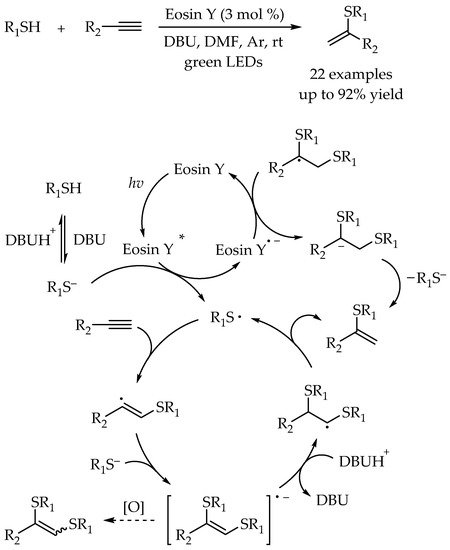
The addition of thiols to alkynes, namely thiol-yne reaction, is also one of the most direct and atom-economic hydrothiolation reaction to construct carbon-sulfur bonds for synthesis of alkenyl thioethers. However, compared to the synthesis of alkyl sulfides, the examples for synthesis of alkenyl sulfides via photocatalytic thiol-yne reactions were less developed. In 2016, The first example of metal-free photoredox thiol-yne reaction under visible-light irradiation using Eosin Y as a cheap and readily available catalyst was reported by Ananikov and co-workers [
47]. Therein, the importance of employing organophotocatalyst is not only to cut the cost, but also to avoid the potential toxic metal contamination. A 3D-printed photoreactor was developed to improve the performance of the catalytic system, and valuable sulfur-functionalized products were obtained in high yields with excellent anti-Markovnikov selectivity. Moreover, the green and efficient synthesis of diene derivatives from industrially available materials demonstrated the potential applicability of this protocol. The reaction mechanism was similar to the previous reported photocatalyzed radical thiol-ene reactions. According to DFT calculations, the high regioselectivity in this reaction was attributed to the fact that the secondary carbon radical generated via anti-Markovnikov addition pathway was more stable than the primary carbon radical (
Scheme 11).
Scheme 11. Visible-light-photocatalyzed thiol-yne reaction by Eosin Y.
In the work reported by Wang’s group in 2019 [
48], when less steric hindered thiols were used in the thiol-yne reaction, the difunctionalization of triple bond could happen, and the corresponding alkyl sulfides could be obtained in good yields. However, it was found that the reaction was hard to stay in the first hydrothiolation stage to obtain the alkenyl thioethers. Even though controlling the usage amounts of thiols, the thiol would still react with the generated alkenyl sulfide in this photocatalytic system, leading to a mixture of alkyl sulfides and alkenyl sulfides (
Scheme 12).
Scheme 12. Visible-light-photocatalyzed thiol-yne reaction by 9-mesityl-10-methylacridinium tetrafluoroborate.
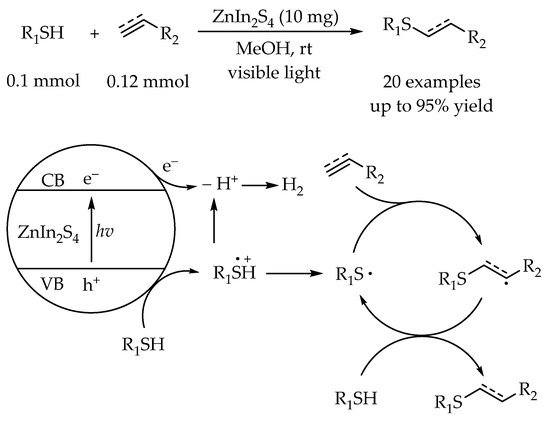
In the thiol-yne reactions, inorganic semiconductor was also proved to be able to act as a suitable and efficient photocatalyst. Li and co-workers developed a visible-light-initiated hydrothiolation of alkenes and alkynes using ZnIn
2S
4 as a photocatalyst in a green solvent [
49]. The high functional group tolerance of this reaction was demonstrated by employing various thiols including aromatic thiols and aliphatic thiols. The ESR (electron spin-resonance spectroscopy) and radical inhibition experiments were conducted to elucidate the mechanism. According to the proposed mechanism, the holes generated after photo-excitation of ZnIn
2S
4 could efficiently oxidize the thiol to form a thiyl radical, then the anti-Markovnikov addition of the thiyl radical to the alkynes/alkenes would generate the alkenyl/alkyl radical for synthesis of the alkenyl sulfides and alkyl sulfides, respectively (
Scheme 13).
Scheme 13. Visible-light-photocatalyzed thiol-ene/yne reaction by ZnIn2S4.
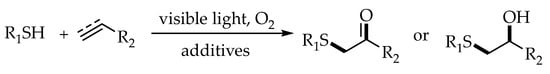
2.3. Photocatalytic Anti-Markovnikov Thiol-Ene/Yne Reactions for Synthesis of β-Hydroxysulfides/β-Keto Sulfides
Introducing another functional group during the hydrothiolation reaction at the α or β-position of thioethers was a brilliant strategy to construct more complex thioethers in one-step process. The key point was how to handle the reactive intermediate, the new generated alkenyl/alkyl radical, which was formed via the thiyl radical addition to the unsaturated hydrocarbons. Based on the previous reports, introducing oxygen in the thiol-ene/yne reactions for further C-O bond formation is a good and efficient choice for synthesis of
β-hydroxysulfides/
β-keto sulfides (
Scheme 14).
Scheme 14. General strategy for synthesis of β-hydroxysulfides/β-ketone sulfides.
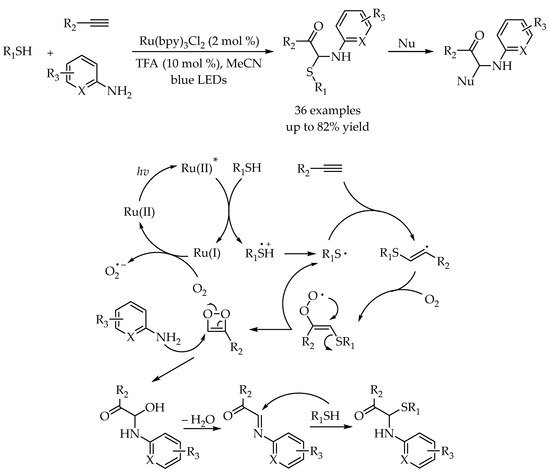
In 2019, Shah and co-workers found that the alkenyl/alkyl radical generated in the radical thiol-yne/ene reaction could undergo oxygen insertion, intermolecular cycloaddition, and nucleophilic addition in the presence of dioxygen and primary arylamine, leading to the construction of α, α-aminothio-substituted carbonyl compounds [
50]. The reactions were carried out under mild reaction conditions with broad substrate scope and excellent efficiency. Furthermore, the low bond dissociation energy of the C-S bond in the products has been exploited for selective functionalization using different nucleophiles to build diverse α, α-disubstituted carbonyl scaffolds (
Scheme 15).
Scheme 15. Visible-light-photocatalyzed gem-difunctionalization of alkynes for the synthesis of α,α-aminothio-substituted carbonyl compounds.
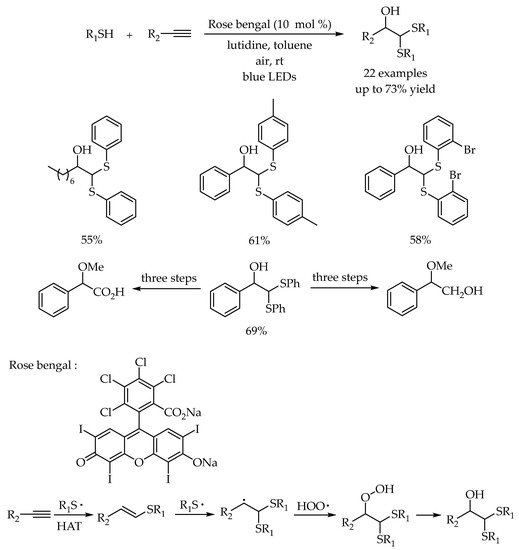
Afterward, the same group expanded this strategy to introduce two sulfur functional group at the α-position and a hydroxy group at the β-position in one-step for the synthesis of β-hydroxydithioacetals [
51]. Starting from the commercially available terminal alkynes and thiophenols, a series of β-hydroxydithioacetals bearing different functional groups were obtained in moderate yields. Further transformation of the product to useful synthons 2-methoxy-2-phenylacetic acid and 2-methoxy-2-phenylethan-1-ol demonstrated the practicability of this photocatalytic system for valuable organic transformations. The reaction mechanism was proposed to proceed via a radical pathway. The anti-Markovnikov addition of thiyl radical to unsaturated hydrocarbons could occur twice to generate a new carbon radical. Then, the radical cross-coupling of the new carbon radical with the hydroperoxide radical and followed by the reduction of the hydroperoxide intermediate produced the corresponding products (
Scheme 16).
Scheme 16. Visible-light photocatalysis for the synthesis of β-hydroxydithioacetals.
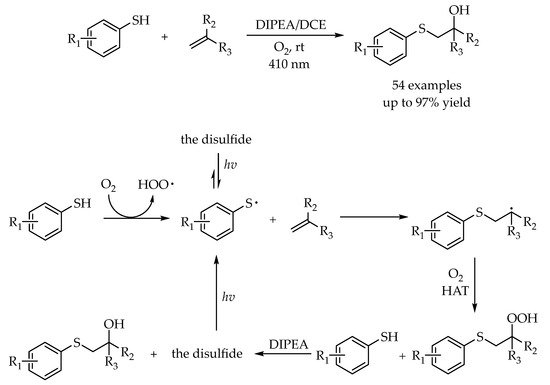
Recently, our group described a visible-light-promoted and tertiary-amine-assisted strategy for efficient hydroxysulfenylation of alkenes with thiophenols to selectively access β-hydroxysulfides in one-step [
52]. Catalyst was unnecessary in this reaction and dioxygen was employed as a green oxidant. Various thiophenols and alkenes could participate in this transformation in good to excellent yields with good functional group tolerance. It is noteworthy that the alkenes with electron-withdrawing groups could also be efficient substrates in this protocol. Moreover, the successful hydroxysulfenylation of structurally complex vinyl-estrone under standard reaction conditions demonstrated that this protocol was a promising strategy for late-stage functionalization of natural products. According to the control experiments and mechanistic investigations, a reasonable reaction pathway was proposed. Under oxygen atmosphere, the thiophenol was first auto-oxidized to generate a thiyl radical, which could attack the alkene to afford an alkyl radical. Then, the generated radical intermediate could quickly react with dioxygen and then followed by a hydrogen atom transfer (HAT) to offer the hydroperoxyl sulfide, which could be in situ reduced directly to the β-hydroxysulfides in the presence of tertiary-amine. Light acting as a driving force played an important role in suppressing the generation of byproduct disulfides and promoted the reaction efficiency (
Scheme 17).
Scheme 17. Light-promoted hydroxysulfenylation of alkenes with thiophenols in the presence of tertiary amines.
2.4. Photocatalytic Anti-Markovnikov Thiol–Ene/Yne Reactions for Synthesis of Sulfoxides
As valuable organic compounds, sulfoxides could be found in many natural and pharmaceutical products. Traditionally, the most direct approach for the synthesis of sulfoxides was the oxidation of thioethers. However, in most cases, two apparent drawbacks were existed in the process. One was the use of excess strong oxidants, which made the reaction usually accompanied with the over oxidation of sulfoxide to sulfone. The other was the multistep operation of the experimental process. Fortunately, the strategy of utilizing tandem thiol-ene reaction cooperated with direct and selective oxidation to sulfoxides was proved to be an efficient alternative (
Scheme 18).
Scheme 18. Strategy for synthesis of sulfoxides via thiol–ene/oxidation tandem reaction.
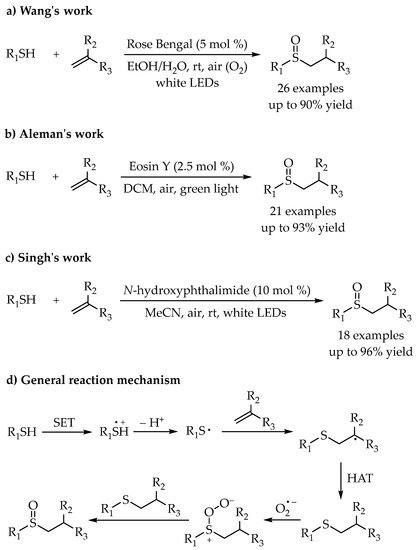
In 2017, Wang’s group described a highly selective synthesis of sulfoxides from alkenes and thiols via visible-light photoredox catalysis using air as the environmentally benign oxidant [
53]. This protocol was compatible with a broad substrate scope, including aryl thiols, aliphatic thiols and the styrenes bearing electron-rich or -poor groups on the aryl rings (
Scheme 19a). Almost at the same time, a similar strategy was reported by Aleman and co-workers [
54]. A photocatalytic synthesis of sulfoxides has been carried out using Eosin Y as a catalyst and atmospheric oxygen as the oxidant. Various alkenes and thiols with different functional groups reacted smoothly under the optimized conditions to obtain the corresponding sulfoxides (
Scheme 19b). The unrecyclable issue of the catalyst in the above two protocols was unavoidable. To address this problem, Singh’s group developed a stable, inexpensive, more efficient, and reusable photocatalyst NHPI, which was suitable for the thiol-ene/oxidation tandem reaction [
55] (
Scheme 19c).The mechanism of those reactions was quite similar. Initially, by visible-light irradiation, a single electron transfer (SET) from thiols to the excited photocatalyst provided a radical cation. After deprotonation, the thiyl radical reacted with alkene leading to the formation of alkyl radical, which would undergo a hydrogen atom transfer (HAT) to offer the thioether intermediate. Finally, a direct photooxidation of the thioethers with dioxygen could give the desired sulfoxides (
Scheme 19d).
Scheme 19. (a–c) Photocatalyzed selective synthesis of sulfoxides via thiol–ene/yne reaction. (d) General reaction mechanism.
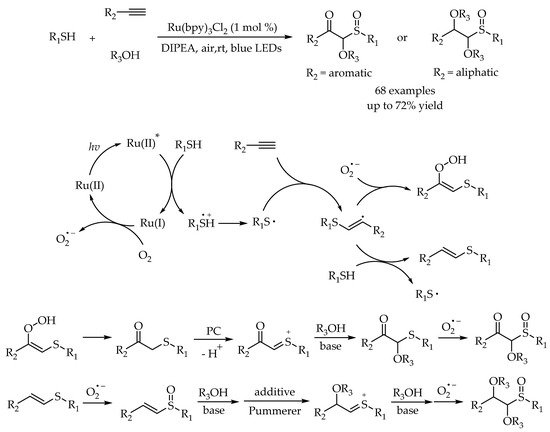
Recently, Shah’s group reported a visible-light photoredox multicomponent reaction for the construction of α-alkoxy-β-ketosulfoxides and α, β-dialkoxysulfoxides using alkynes, thiols, and alcohols as starting materials [
56]. This elegant protocol provided a solution to access α-substituted sulfoxides in one-step synthesis. Substrates with various functional groups were available in this transformation, giving rise to the α-substituted sulfoxides in good yields. Based on the control experiments and the previous literatures, a plausible reaction mechanism was proposed. Generally, after the formation of thiyl radical, the radical addition of the thiyl radical to the terminal alkyne could afford the vinyl sulfide intermediate, which could undergo the oxidation and nucleophilic addition continuously to furnish the corresponding products (
Scheme 20).
Scheme 20. The general mechanism of photoredox synthesis of sulfoxides.