 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Aamir Rana | + 1811 word(s) | 1811 | 2022-01-27 11:43:52 | | | |
| 2 | Yvaine Wei | Meta information modification | 1811 | 2022-02-09 02:31:56 | | |
Video Upload Options
The constitutively active BCR-ABL1 tyrosine kinase, found in t(9;22)(q34;q11) chromosomal translocation-derived leukemia, initiates an extremely complex signaling transduction cascade that induces a strong state of resistance to chemotherapy. Targeted therapies based on tyrosine kinase inhibitors (TKIs), such as imatinib, dasatinib, nilotinib, bosutinib, and ponatinib, have revolutionized the treatment of BCR-ABL1-driven leukemia, particularly chronic myeloid leukemia (CML). However, TKIs do not cure CML patients, as some develop TKI resistance and the majority relapse upon withdrawal from treatment. Importantly, although BCR-ABL1 tyrosine kinase is necessary to initiate and establish the malignant phenotype of Ph-related leukemia, in the later advanced phase of the disease, BCR-ABL1-independent mechanisms are also in place.
1. Introduction
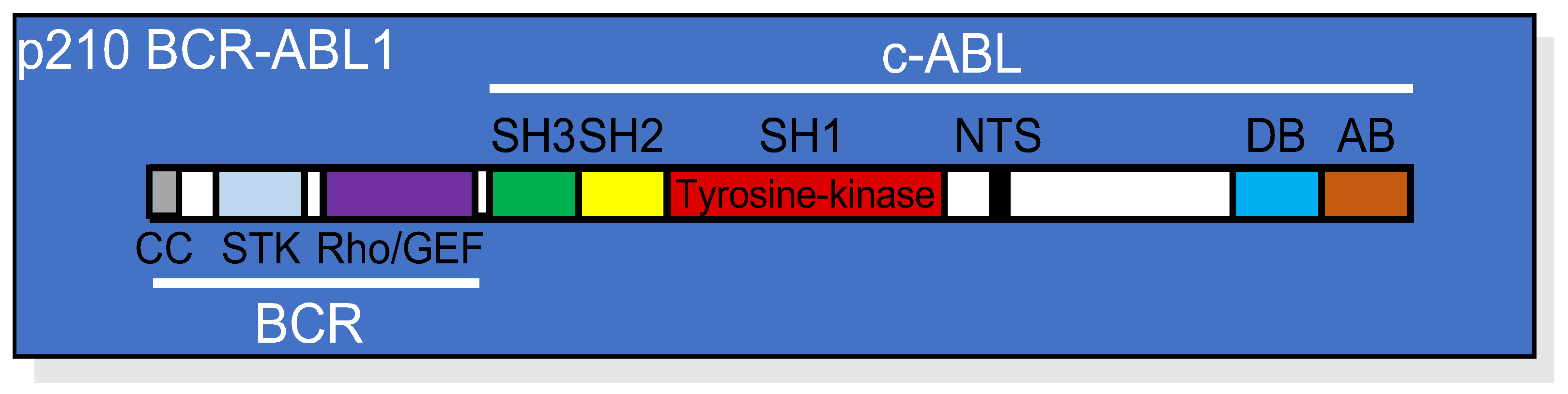
2. BCR-ABL1 Tyrosine Kinase-Dependent Signaling Cascade
2.1. BCR-ABL1 Activation of the RAS/RAF/MAPK Pathway
2.2. BCR-ABL1 Activation of the PI3K/AKT/mTOR Pathway
2.3. BCR-ABL1 Activation of the JAK/STAT Pathway
2.4. BCR-ABL1 Activation of the WNT/β-Catenin Pathway
2.5. BCR-ABL1 Activation of the PP2A Pathway
3. BCR-ABL1 Kinase-Independent Alternative Survival Signals
4. Resistance to Apoptosis in CML
5. Tyrosine Kinase Inhibitors and the Paradigm Shift of CML Treatment
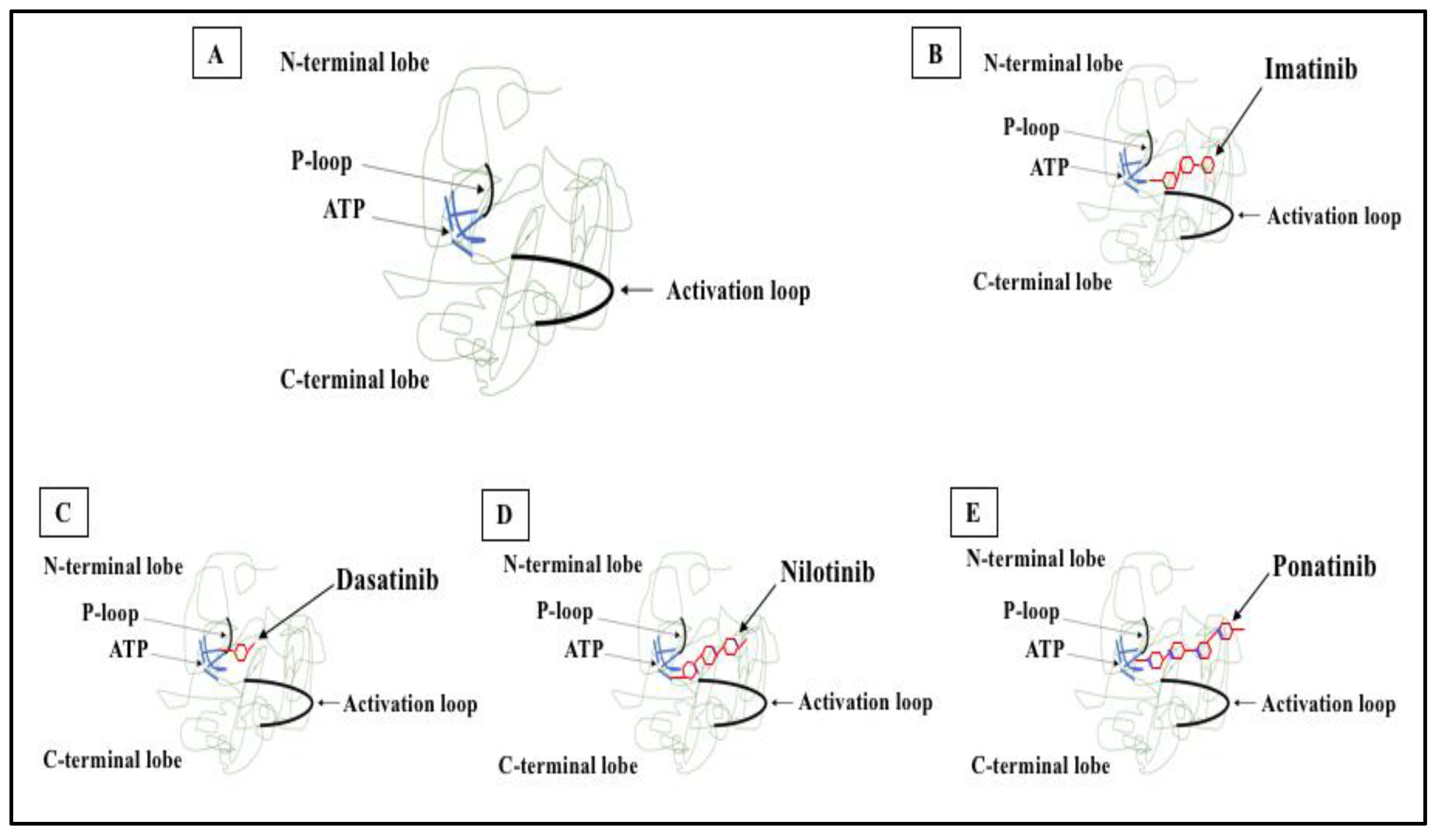
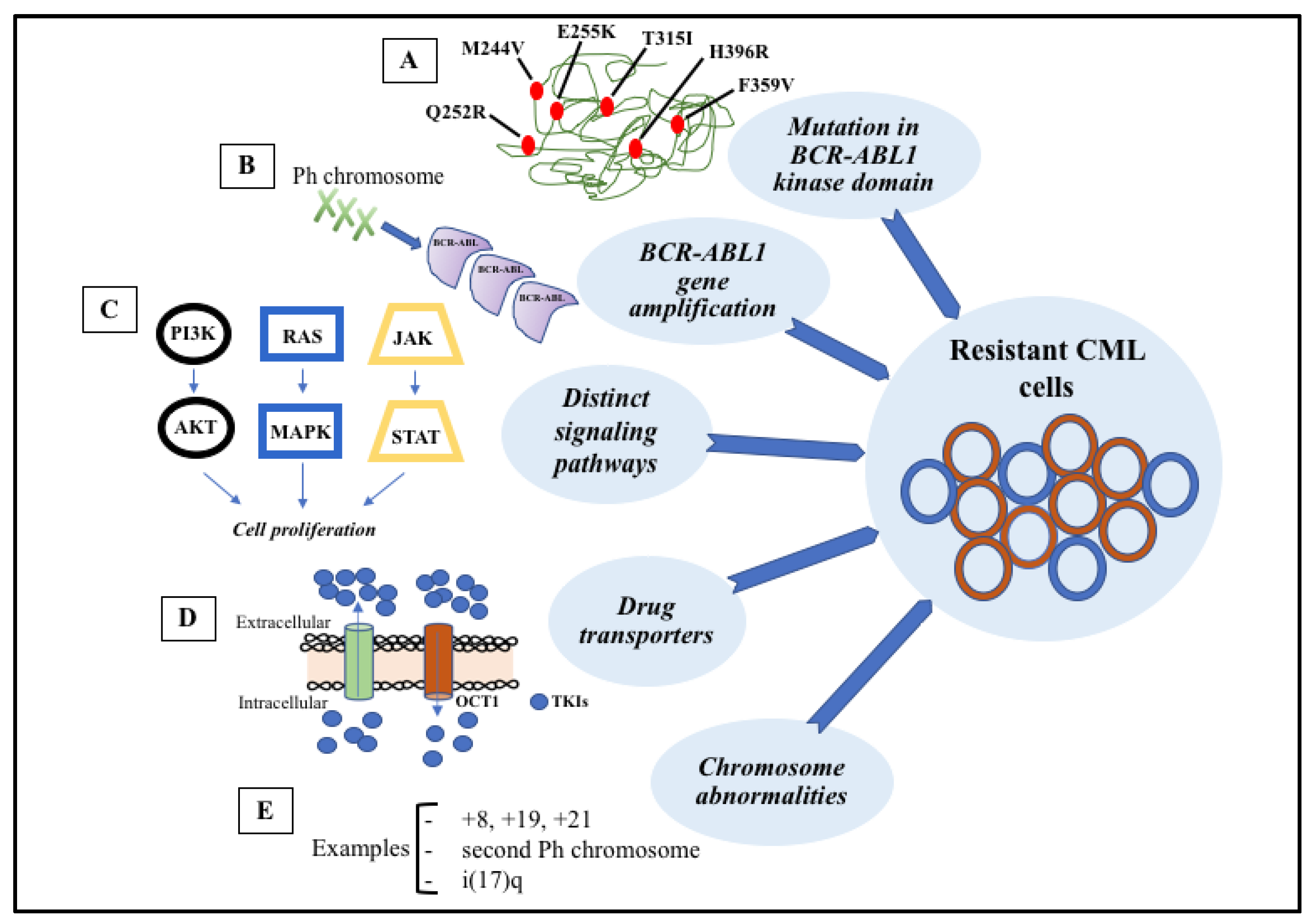
6. Mechanisms of Resistance to TKI
References
- Nowell, P.C.; Hungerford, D.A. Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109.
- Pane, F.; Frigeri, F.; Sindona, M.; Luciano, L.; Ferrara, F.; Cimino, R.; Meloni, G.; Saglio, G.; Salvatore, F.; Rotoli, B. Neutrophilic-Chronic Myeloid Leukemia: A Distinct Disease with a Specific Molecular Marker (BCR/ABL With C3/A2 Junction). Blood 1996, 88, 2410–2414.
- Walker, L.C.; Ganesan, T.S.; Dhut, S.; Gibbons, B.; Andrew Lister, T.; Rothbardt, J.; Young, B.D. Novel Chimaeric Protein Expressed in Philadelphia Positive Acute Lymphoblastic Leukaemia. Nature 1987, 329, 851–853.
- Heisterkamp, N.; Stephenson·, J.R.; Groffen·, J.; Hansent, P.F.; de Klein, A.; Bartram, C.R.; Grosveld, G. Localization of the C-Abl Oncogene Adjacent to a Translocation Break Point in Chronic Myelocytic Leukaemia. Nature 1983, 306, 239–242.
- Mcwhirter, J.R.; Wang, J.Y.J. An Actin-Binding Function Contributes to Transformation by the Bcr-Abl Oncoprotein of Philadelphia Chromosome-Positive Human Leukemias. EMBO J. 1993, 12, 1533–1546.
- Peiris, M.N.; Li, F.; Donoghue, D.J. BCR: A promiscuous fusion partner in hematopoietic disorders. Oncotarget 2019, 10, 2738–2754.
- Pendergast, A.M.; Quilliam, L.A.; Cripe, L.D.; Bassing, C.H.; Dai, Z.; Li, N.; Batzer, A.; Rabun, K.M.; Der, C.J.; Schlessinger, J.; et al. BCR-ABL-Induced Oncogenesis Is Mediated by Direct Interaction with the SH2 Domain of the GRB-2 Adaptor Protein. Cell 1993, 75, 175–785.
- Van Etten, R.A.; Jackson, P.; Baltimore, D. The Mouse Type IV C-Abl Gene Product Is a Nuclear Protein, and Activation of Transforming Ability Is Associated with Cytoplasmic Localization. Cell 1989, 58, 669–678.
- Brehme, M.; Hantschel, O.; Colinge, J.; Kaupe, I.; Planyavsky, M.; Kö Cher, T.; Mechtler, K.; Bennett, K.L.; Superti-Furga, G. Charting the Molecular Network of the Drug Target Bcr-Abl. Proc. Natl. Acad. Sci. USA 2009, 106, 7414–7419.
- Sattler, M.; Golam Mohi, M.; Pride, Y.B.; Quinnan, L.R.; Malouf, N.A.; Podar, K.; Gesbert, F.; Iwasaki, H.; Li, S.; van Etten, R.A.; et al. Critical Role for Gab2 in Transformation by BCR/ABL. Cancer Cell 2002, 1, 479–492.
- Gregory, M.A.; Phang, T.L.; Neviani, P.; Alvarez-Calderon, F.; Eide, C.A.; O’Hare, T.; Zaberezhnyy, V.; Williams, R.T.; Druker, B.J.; Perrotti, D.; et al. Wnt/Ca2+/NFAT Signaling Maintains Survival of Ph+ Leukemia Cells upon Inhibition of Bcr-Abl. Cancer Cell 2010, 18, 74–87.
- Hu, Y.; Chen, Y.; Douglas, L.; Li, S. β-Catenin Is Essential for Survival of Leukemic Stem Cells Insensitive to Kinase Inhibition in Mice with BCR-ABL-Induced Chronic Myeloid Leukemia. Leukemia 2009, 23, 109–116.
- Reins, J.; Mossner, M.; Neumann, M.; Platzbecker, U.; Schumann, C.; Thiel, E.; Hofmann, W.K. Transcriptional Down-Regulation of the Wnt Antagonist SFRP1 in Haematopoietic Cells of Patients with Different Risk Types of MDS. Leuk. Res. 2010, 34, 1610–1616.
- Danisz, K.; Blasiak, J. Role of Anti-Apoptotic Pathways Activated by BCR/ABL in the Resistance of Chronic Myeloid Leukemia Cells to Tyrosine Kinase Inhibitors. Acta Biochim. Pol. 2013, 4, 503–514.
- Ren, R. Mechanisms of BCR-ABL in the Pathogenesis of Chronic Myelogenous Leukaemia. Nat. Rev. Cancer 2005, 5, 172–183.
- Hochhaus, A.; Kreil, S.; Corbin, A.S.; la Rosée, P.; Müller, M.C.; Lahaye, T.; Hanfstein, B.; Schoch, C.; Cross, N.C.P.; Berger, U.; et al. Molecular and Chromosomal Mechanisms of Resistance to Imatinib (STI571) Therapy. Leukemia 2002, 16, 2190–2196.
- Dinner, S.; Platanias, L.C. Targeting the MTOR Pathway in Leukemia. J. Cell. Biochem. 2016, 117, 1745–1752.
- Ilaria, R.L.; van Etten, R.A. P210 and P190 BCR/ABL Induce the Tyrosine Phosphorylation and DNA Binding Activity of Multiple Specific STAT Family Members. J. Biol. Biochem. 1996, 271, 6188–6195.
- Xie, S.; Wang, Y.; Liu, J.; Sun, T.; Wilson, M.B.; Smithgall, T.E.; Arlinghaus, R.B. Involvement of Jak2 Tyrosine Phosphorylation in Bcr ± Abl Transformation. Oncogene 2001, 20, 6188–6195.
- Hoelbl, A.; Schuster, C.; Kovacic, B.; Zhu, B.; Wickre, M.; Hoelzl, M.A.; Fajmann, S.; Grebien, F.; Warsch, W.; Stengl, G.; et al. Stat5 Is Indispensable for the Maintenance of Bcr/Abl-Positive Leukaemia. EMBO Mol. Med. 2010, 2, 98–110.
- Gallipoli, P.; Cook, A.; Rhodes, S.; Hopcroft, L.; Wheadon, H.; Whetton, A.D.; Jørgensen, H.G.; Bhatia, R.; Holyoake, T.L. JAK2/STAT5 Inhibition by Nilotinib with Ruxolitinib Contributes to the Elimination of CML CD34 + Cells in Vitro and in Vivo. Blood 2014, 124, 1492–1501.
- Chai, S.K.; Nichols, G.L.; Rothman, P. Constitutive activation of JAKs and STATs in BCR-Abl-expressing cell lines and peripheral blood cells derived from leukemic patients. J. Immunol. 2021, 159, 4720–4728.
- Fetisov, T.I.; Lesovaya, E.A.; Yakubovskaya, M.G.; Kirsanov, K.I.; Belitsky, G.A. Alterations in WNT Signaling in Leukemias. Biochemistry 2018, 83, 1448–1458.
- Soares-Lima, S.C.; Pombo-de-Oliveira, M.S.; Carneiro, F.R.G. The Multiple Ways Wnt Signaling Contributes to Acute Leukemia Pathogenesis. J. Leukoc. Biol. 2020, 108, 1081–1099.
- Janssens, V.; Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001, 353, 417–439.
- Gregor, T.; Bosakova, M.K.; Nita, A.; Abraham, S.P.; Fafilek, B.; Cernohorsky, N.H.; Rynes, J.; Foldynova-Trantirkova, S.; Zackova, D.; Mayer, J.; et al. Elucidation of Protein Interactions Necessary for the Maintenance of the BCR–ABL Signaling Complex. Cell. Mol. Life Sci. 2020, 77, 3885–3903.
- Bueno-da-Silva, A.E.B.; Brumatti, G.; Russo, F.O.; Green, D.R.; Amarante-Mendes, G.P. Bcr-Abl-Mediated Resistance to Apoptosis Is Independent of Constant Tyrosine-Kinase Activity. Cell Death Differ. 2003, 10, 592–598.
- Raitano, A.B.; Whang, Y.E.; Sawyers, C.L. Signal Transduction by Wild-Type and Leukemogenic Abl Proteins. Biochim. Biophys. Acta 1997, 1333, F201–F216.
- Sattler, M.; Salgia, R. Activation of Hematopoietic Growth Factor Signal Transduction Pathways by the Human Oncogene BCR/ABL. Cytokine Growth Factor Rev. 1997, 8, 63–79.
- Zou, X.; Calame, K. Signaling Pathways Activated by Oncogenic Forms of Abl Tyrosine Kinase. J. Biol. Chem. 1999, 274, 18141–18144.
- Dormer, P.; Lau, B.; Wilmanns, W. Kinetics of bone marrow cell production in human acute and chronic myeloid leukemias. Leuk. Res. 1980, 4, 231–237.
- Cortez, D.; Kadlec, L.; Pendergast, A.M. Structural and Signaling Requirements for BCR-ABL-Mediated Transformation and Inhibition of Apoptosis. Mol. Cell. Biol. 1995, 15, 5531–5541.
- Bedi, A.; Zehnbauer, B.A.; Barber, J.P.; Sharkis, S.J.; Jones, R.J. Inhibition of apoptosis by BCR-ABL in chronic myeloid leukemia. Blood 1994, 83, 2038–2044.
- Mcgahon, A.J.; Brown, D.G.; Martin, S.J.; Amarante-Mendes, G.P.; Cotter, T.G.; Cohen, G.M.; Green, D.R. Downregulation of Bcr-Abl in K562 Cells Restores Susceptibility to Apoptosis: Characterization of the Apoptotic Death. Cell Death Differ. 1997, 4, 95–104.
- Amarante-Mendes, G.P.; Finucane, D.M.; Martin, S.J.; Cotter, T.G.; Salvesen, G.S.; Green, D.R. Anti-Apoptotic Oncogenes Prevent Caspase-Dependent and Independent Commitment for Cell Death. Cell Death Differ. 1998, 5, 298–306.
- Fernandez-Luna, J.L. Bcr-Abl and Inhibition of Apoptosis in Chronic Myelogenous Leukemia Cells. Apoptosis 2000, 5, 315–318.
- Brumatti, G.; Weinlich, R.; Chehab, C.F.; Yon, M.; Amarante-Mendes, G.P. Comparison of the Anti-Apoptotic Effects of Bcr-Abl, Bcl-2 and Bcl-XL Following Diverse Apoptogenic Stimuli. FEBS Lett. 2003, 541, 57–63.
- Ghaffari, S.; Jagani, Z.; Kitidis, C.; Lodish, H.F.; Khosravi-Far, R. Cytokines and BCR-ABL Mediate Suppression of TRAIL-Induced Apoptosis through Inhibition of Forkhead FOXO3a Transcription Factor. Proc. Natl. Acad. Sci. USA 2003, 100, 6523–6528.
- Lugo, T.G.; Pendergast, A.M.; Muller, A.J.; Witte, O.N. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science 1990, 247, 1079–1082.
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 Recommendations for Treating Chronic Myeloid Leukemia. Leukemia 2020, 34, 966–984.
- Hehlmann, R. The New Eln Recommendations for Treating Cml. J. Clin. Med. 2020, 9, 3671.
- Khorashad, J.S.; Kelley, T.W.; Szankasi, P.; Mason, C.C.; Soverini, S.; Adrian, L.T.; Eide, C.A.; Zabriskie, M.S.; Lange, T.; Estrada, J.C.; et al. BCR-ABL1 Compound Mutations in Tyrosine Kinase Inhibitor-Resistant CML: Frequency and Clonal Relationships. Blood 2013, 121, 489–498.
- Soverini, S.; Branford, S.; Nicolini, F.E.; Talpaz, M.; Deininger, M.W.N.; Martinelli, G.; Müller, M.C.; Radich, J.P.; Shah, N.P. Implications of BCR-ABL1 Kinase Domain-Mediated Resistance in Chronic Myeloid Leukemia. Leuk. Res. 2014, 38, 10–20.
- Eck, M.J.; Manley, P.W. The Interplay of Structural Information and Functional Studies in Kinase Drug Design: Insights from BCR-Abl. Curr. Opin. Cell Biol. 2009, 21, 288–295.

