The constitutively active BCR-ABL1 tyrosine kinase, found in t(9;22)(q34;q11) chromosomal translocation-derived leukemia, initiates an extremely complex signaling transduction cascade that induces a strong state of resistance to chemotherapy. Targeted therapies based on tyrosine kinase inhibitors (TKIs), such as imatinib, dasatinib, nilotinib, bosutinib, and ponatinib, have revolutionized the treatment of BCR-ABL1-driven leukemia, particularly chronic myeloid leukemia (CML). However, TKIs do not cure CML patients, as some develop TKI resistance and the majority relapse upon withdrawal from treatment. Importantly, although BCR-ABL1 tyrosine kinase is necessary to initiate and establish the malignant phenotype of Ph-related leukemia, in the later advanced phase of the disease, BCR-ABL1-independent mechanisms are also in place.
- BCR-ABL1
- chronic myeloid leukemia
- tyrosine kinase inhibitors
1. Introduction
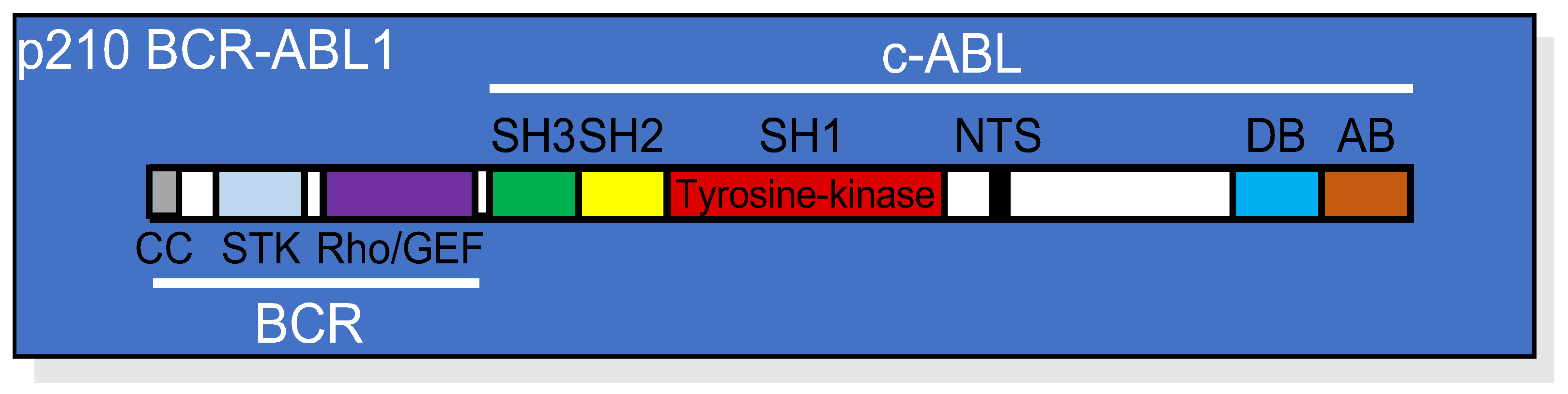
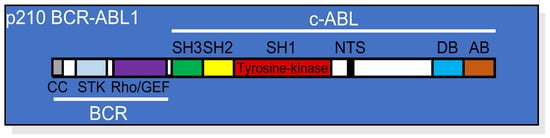 Figure 1. Linear structure of p210 BCR-ABL1 showing the relative position of each domain from both the BCR and the c-ABL portions of the protein. CC, coil-coiled oligomerization domain; STK, serine/threonine kinase domain; Rho/GEF domain; SH, (SRC homology domains) 1, 2, and 3; NTS, nuclear translocation signal; DB, DNA binding domain; AB, actin binding domain.
Figure 1. Linear structure of p210 BCR-ABL1 showing the relative position of each domain from both the BCR and the c-ABL portions of the protein. CC, coil-coiled oligomerization domain; STK, serine/threonine kinase domain; Rho/GEF domain; SH, (SRC homology domains) 1, 2, and 3; NTS, nuclear translocation signal; DB, DNA binding domain; AB, actin binding domain.2. BCR-ABL1 Tyrosine Kinase-Dependent Signaling Cascade
2.1. BCR-ABL1 Activation of the RAS/RAF/MAPK Pathway
2.2. BCR-ABL1 Activation of the PI3K/AKT/mTOR Pathway
BCR-ABL1 activates the PI3K/AKT/mTOR pathway both directly and indirectly through the induction of autocrine cytokines [21][14]. The interaction between BCR-ABL1 and PI3K can occur via GRB2, GAB-2, SHC, c-CBL, and CRKL [15,22,23][10][15][16]. AKT also induces the downstream activation of mammalian target of rapamycin (mTOR), which works as a catalytic subunit of the mTORC1 and mTORC2 protein complexes [31][17].2.3. BCR-ABL1 Activation of the JAK/STAT Pathway
Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling is involved in growth factor independence and resistance to apoptosis in CML [43,44,45,46,47][18][19][20][21][22]. JAK2, STAT1, STAT3, and STAT5 were shown to be constitutively active in CML cell lines [47][22].2.4. BCR-ABL1 Activation of the WNT/β-Catenin Pathway
A disturbance in the canonical WNT/β-catenin signaling pathway is associated with the pathogenesis of leukemia [53,54][23][24]. In the absence of WNT activation, β-catenin is phosphorylated and ubiquitinated in the context of a multiprotein complex composed of glycogen synthase kinase 3 (GSK-3), creatin kinase 1 (CK1), axis inhibitor (Axin), adenomatous polyposis coli (APC), protein phosphatase 2A (PP2A), and the E3-ubiquitin ligase β-transducin repeat-containing protein (β-TrCP), followed by further proteasomal degradation [53,54][23][24].2.5. BCR-ABL1 Activation of the PP2A Pathway
PP2A is a tumor suppressor serine-threonine phosphatase that negatively regulates the mitogenic and survival signals emanating from PI3K/AKT, RAS/MAPK, and MYC pathways [59][25].3. BCR-ABL1 Kinase-Independent Alternative Survival Signals
Unquestionably, the BCR-ABL1 tyrosine kinase-dependent signaling events are required for the transformation of Ph1 chromosome-positive leukemia. However, the resistance to TKIs observed in some CML patients suggests that signals emanating from the BCR-ABL1 protein independently of its tyrosine kinase activity may take over, allowing the survival of leukemic cells and relapse of the disease. Indeed, it was recently shown that inhibition of the catalytic activity does not completely dismantle the BCR-ABL1 molecular complex [63][26]. Signaling proteins, such as p85α-PI3K, GRB2, SHIP2, SHC1, SOS1, and c-CBL, remain associated with BCR-ABL1, whereas CRK, CRKL, or GAB2 seem to detach from the complex [63][26]. Therefore, residual signaling transduction events appear to be sufficient to maintain survival of CML cells in the absence of tyrosine kinase activity [64][27].4. Resistance to Apoptosis in CML
BCR-ABL1 activates multiple signaling pathways to induce leukemogenesis, which results in growth factor-independency and regulation of adhesion and invasion [71,72,73][28][29][30]. On the other hand, BCR-ABL1-positive cells generally display normal mitotic indices and do not show increased overall proliferation [74][31]. Perhaps the most notable aspect of BCR-ABL1-mediated leukemogenesis is the vigorous state of resistance to apoptosis that is conferred to the transformed cells [19,75,76,77,78,79,80][32][33][34][35][36][37][38]. The enforced expression of BCR-ABL1 in hematopoietic cell lineages revealed its potential to prevent apoptosis induced by a variety of stimuli, including growth factor withdrawal, γ-irradiation, death receptor agonists, and multiple chemotherapeutic drugs [19,75,76,77,78,79][32][33][34][35][36][37]. Studies with point mutations at the autophosphorylation site (Y793F), the phosphotyrosine binding motif (R552L), and/or at the GRB2-binding site (Y177F) demonstrated that BCR-ABL1-mediated resistance to apoptosis depends on the cellular context. For instance, enforced expression of a BCR-ABL1-Y177F/R552L/Y793F triple mutant in IL-3-dependent lymphoblastoid 32D murine cells did not confer IL-3 independency or resistance to γ-irradiation-induced apoptosis; however, the same mutant protected BaF3 cells, a different IL-3-dependent pro-B murine cell line, from these apoptogenic stimuli [19][32]. Moreover, enforced expression of wild type or the above-mentioned BCR-ABL1 mutants protected the apoptosis-sensitive human acute promyelocytic leukemia HL-60 cell line from a variety of apoptogenic insults to the same extent [77][35]. Therefore, different cellular contexts may provide alternative pathways that contribute to the survival of BCR-ABL1-positive cells.5. Tyrosine Kinase Inhibitors and the Paradigm Shift of CML Treatment
The discovery that constitutive BCR-ABL1 tyrosine kinase activity was crucial for the development of CML [96][39] warranted a TK-targeting therapeutic strategy. Consequently, several TKIs were developed to target the ATP binding site of the kinase domain, thereby preventing phosphorylation of the target protein and subsequent signaling events (Figure 2). Imatinib, nilotinib, dasatinib, bosutinib, ponatinib, and asciminib are currently used for the treatment of CML and are briefly described below.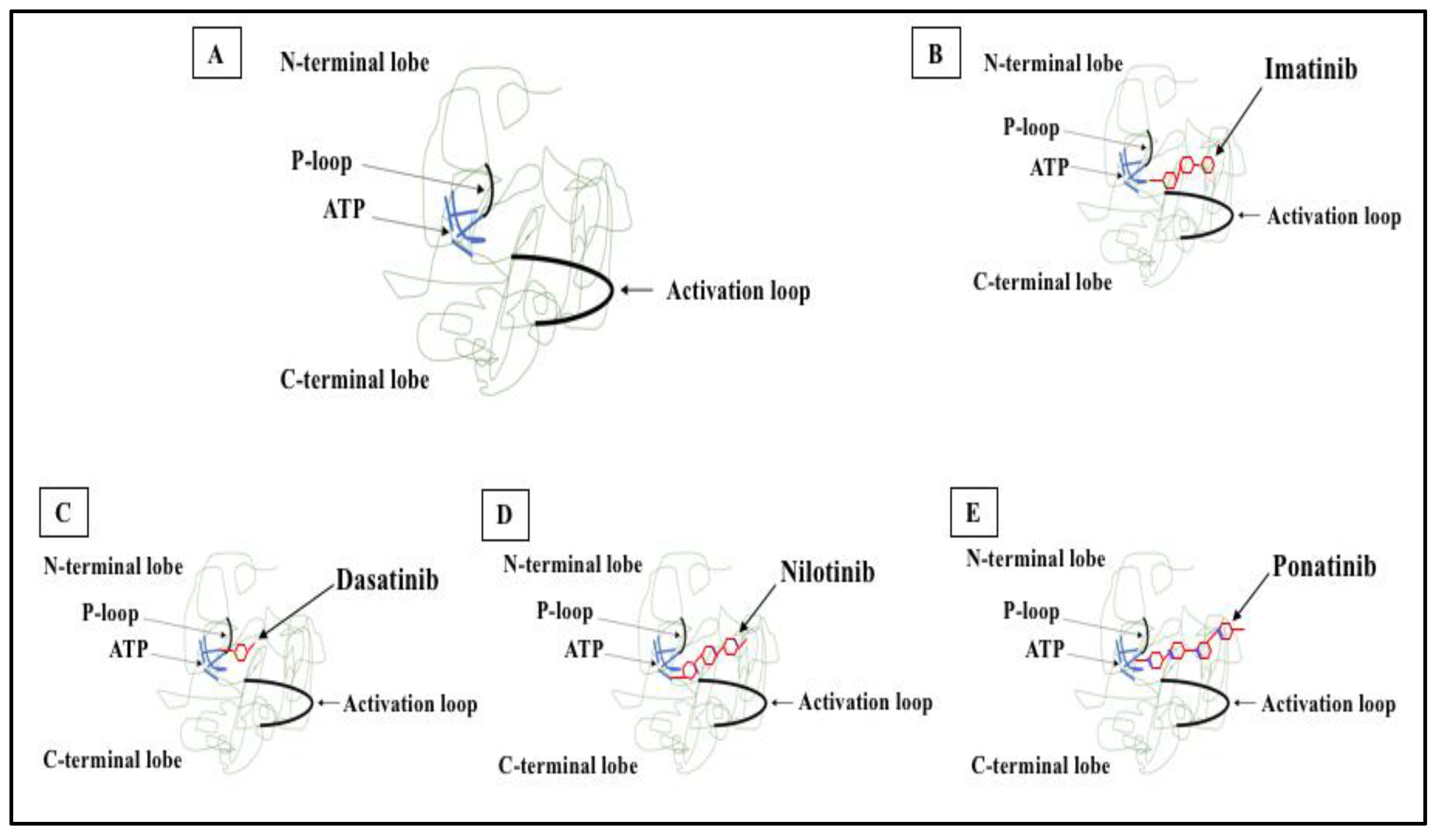
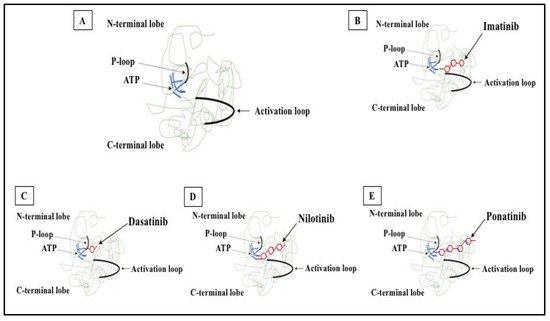 Figure 2. 2D structure of BCR-ABL1 kinase domain and binding sites for TKIs. (A) BCR-ABL1 is a constitutively active kinase that binds ATP and transfers a phosphate from ATP to tyrosine residues on various substrates. This activates downstream signaling pathways, leading to abnormal cellular adhesion and proliferation of myeloid cells and inhibition of apoptosis. TKIs were developed to specifically block the binding of ATP to the BCR-ABL tyrosine kinase, inactivating the constitutive tyrosine kinase activity and inhibiting downstream pathways. (B) Imatinib (first generation TKI) binds to the BCR-ABL kinase domain in its inactive conformation through the ATP binding site. (C) Dasatinib (second generation TKI) inhibits the BCR-ABL tyrosine kinase performance at the ATP site in ABL regardless of protein conformation (active or inactive). (D) Nilotinib (second generation TKI) connects to an inactive conformation of the BCR-ABL protein, taking an analogous region that would be occupied by ATP. (E) Ponatinib (third generation TKI) has multiple contact points for the inactive conformation of the ABL and for the T315I mutation.
Figure 2. 2D structure of BCR-ABL1 kinase domain and binding sites for TKIs. (A) BCR-ABL1 is a constitutively active kinase that binds ATP and transfers a phosphate from ATP to tyrosine residues on various substrates. This activates downstream signaling pathways, leading to abnormal cellular adhesion and proliferation of myeloid cells and inhibition of apoptosis. TKIs were developed to specifically block the binding of ATP to the BCR-ABL tyrosine kinase, inactivating the constitutive tyrosine kinase activity and inhibiting downstream pathways. (B) Imatinib (first generation TKI) binds to the BCR-ABL kinase domain in its inactive conformation through the ATP binding site. (C) Dasatinib (second generation TKI) inhibits the BCR-ABL tyrosine kinase performance at the ATP site in ABL regardless of protein conformation (active or inactive). (D) Nilotinib (second generation TKI) connects to an inactive conformation of the BCR-ABL protein, taking an analogous region that would be occupied by ATP. (E) Ponatinib (third generation TKI) has multiple contact points for the inactive conformation of the ABL and for the T315I mutation.6. Mechanisms of Resistance to TKI
 Figure 3. Signaling pathways involved in the development of target therapy resistance. (A) Molecular structure of BCR-ABL1 kinase domain with some mutations (indicated in red). (B) Gene amplification can lead to overproduction of tyrosine kinase. (C) Constitutive activation of signaling pathways, such as PI3K-AKT, RAS-MAPK, and JAK-STAT, result in cell proliferation and anti-apoptotic mechanisms. (D) Intracellular concentrations of TKIs can be modified through membrane transporters that may cause increased efflux or decreased influx. (E) The most common chromosome abnormalities involved in karyotype evolution are trisomy 8, trisomy 19, trisomy 21, second Ph chromosome, and isochromosome 17.
Figure 3. Signaling pathways involved in the development of target therapy resistance. (A) Molecular structure of BCR-ABL1 kinase domain with some mutations (indicated in red). (B) Gene amplification can lead to overproduction of tyrosine kinase. (C) Constitutive activation of signaling pathways, such as PI3K-AKT, RAS-MAPK, and JAK-STAT, result in cell proliferation and anti-apoptotic mechanisms. (D) Intracellular concentrations of TKIs can be modified through membrane transporters that may cause increased efflux or decreased influx. (E) The most common chromosome abnormalities involved in karyotype evolution are trisomy 8, trisomy 19, trisomy 21, second Ph chromosome, and isochromosome 17.References
- Nowell, P.C.; Hungerford, D.A. Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109.
- Pane, F.; Frigeri, F.; Sindona, M.; Luciano, L.; Ferrara, F.; Cimino, R.; Meloni, G.; Saglio, G.; Salvatore, F.; Rotoli, B. Neutrophilic-Chronic Myeloid Leukemia: A Distinct Disease with a Specific Molecular Marker (BCR/ABL With C3/A2 Junction). Blood 1996, 88, 2410–2414.
- Walker, L.C.; Ganesan, T.S.; Dhut, S.; Gibbons, B.; Andrew Lister, T.; Rothbardt, J.; Young, B.D. Novel Chimaeric Protein Expressed in Philadelphia Positive Acute Lymphoblastic Leukaemia. Nature 1987, 329, 851–853.
- Heisterkamp, N.; Stephenson·, J.R.; Groffen·, J.; Hansent, P.F.; de Klein, A.; Bartram, C.R.; Grosveld, G. Localization of the C-Abl Oncogene Adjacent to a Translocation Break Point in Chronic Myelocytic Leukaemia. Nature 1983, 306, 239–242.
- Mcwhirter, J.R.; Wang, J.Y.J. An Actin-Binding Function Contributes to Transformation by the Bcr-Abl Oncoprotein of Philadelphia Chromosome-Positive Human Leukemias. EMBO J. 1993, 12, 1533–1546.
- Peiris, M.N.; Li, F.; Donoghue, D.J. BCR: A promiscuous fusion partner in hematopoietic disorders. Oncotarget 2019, 10, 2738–2754.
- Pendergast, A.M.; Quilliam, L.A.; Cripe, L.D.; Bassing, C.H.; Dai, Z.; Li, N.; Batzer, A.; Rabun, K.M.; Der, C.J.; Schlessinger, J.; et al. BCR-ABL-Induced Oncogenesis Is Mediated by Direct Interaction with the SH2 Domain of the GRB-2 Adaptor Protein. Cell 1993, 75, 175–785.
- Van Etten, R.A.; Jackson, P.; Baltimore, D. The Mouse Type IV C-Abl Gene Product Is a Nuclear Protein, and Activation of Transforming Ability Is Associated with Cytoplasmic Localization. Cell 1989, 58, 669–678.
- Brehme, M.; Hantschel, O.; Colinge, J.; Kaupe, I.; Planyavsky, M.; Kö Cher, T.; Mechtler, K.; Bennett, K.L.; Superti-Furga, G. Charting the Molecular Network of the Drug Target Bcr-Abl. Proc. Natl. Acad. Sci. USA 2009, 106, 7414–7419.
- Sattler, M.; Golam Mohi, M.; Pride, Y.B.; Quinnan, L.R.; Malouf, N.A.; Podar, K.; Gesbert, F.; Iwasaki, H.; Li, S.; van Etten, R.A.; et al. Critical Role for Gab2 in Transformation by BCR/ABL. Cancer Cell 2002, 1, 479–492.
- Gregory, M.A.; Phang, T.L.; Neviani, P.; Alvarez-Calderon, F.; Eide, C.A.; O’Hare, T.; Zaberezhnyy, V.; Williams, R.T.; Druker, B.J.; Perrotti, D.; et al. Wnt/Ca2+/NFAT Signaling Maintains Survival of Ph+ Leukemia Cells upon Inhibition of Bcr-Abl. Cancer Cell 2010, 18, 74–87.
- Hu, Y.; Chen, Y.; Douglas, L.; Li, S. β-Catenin Is Essential for Survival of Leukemic Stem Cells Insensitive to Kinase Inhibition in Mice with BCR-ABL-Induced Chronic Myeloid Leukemia. Leukemia 2009, 23, 109–116.
- Reins, J.; Mossner, M.; Neumann, M.; Platzbecker, U.; Schumann, C.; Thiel, E.; Hofmann, W.K. Transcriptional Down-Regulation of the Wnt Antagonist SFRP1 in Haematopoietic Cells of Patients with Different Risk Types of MDS. Leuk. Res. 2010, 34, 1610–1616.
- Danisz, K.; Blasiak, J. Role of Anti-Apoptotic Pathways Activated by BCR/ABL in the Resistance of Chronic Myeloid Leukemia Cells to Tyrosine Kinase Inhibitors. Acta Biochim. Pol. 2013, 4, 503–514.
- Ren, R. Mechanisms of BCR-ABL in the Pathogenesis of Chronic Myelogenous Leukaemia. Nat. Rev. Cancer 2005, 5, 172–183.
- Hochhaus, A.; Kreil, S.; Corbin, A.S.; la Rosée, P.; Müller, M.C.; Lahaye, T.; Hanfstein, B.; Schoch, C.; Cross, N.C.P.; Berger, U.; et al. Molecular and Chromosomal Mechanisms of Resistance to Imatinib (STI571) Therapy. Leukemia 2002, 16, 2190–2196.
- Dinner, S.; Platanias, L.C. Targeting the MTOR Pathway in Leukemia. J. Cell. Biochem. 2016, 117, 1745–1752.
- Ilaria, R.L.; van Etten, R.A. P210 and P190 BCR/ABL Induce the Tyrosine Phosphorylation and DNA Binding Activity of Multiple Specific STAT Family Members. J. Biol. Biochem. 1996, 271, 6188–6195.
- Xie, S.; Wang, Y.; Liu, J.; Sun, T.; Wilson, M.B.; Smithgall, T.E.; Arlinghaus, R.B. Involvement of Jak2 Tyrosine Phosphorylation in Bcr ± Abl Transformation. Oncogene 2001, 20, 6188–6195.
- Hoelbl, A.; Schuster, C.; Kovacic, B.; Zhu, B.; Wickre, M.; Hoelzl, M.A.; Fajmann, S.; Grebien, F.; Warsch, W.; Stengl, G.; et al. Stat5 Is Indispensable for the Maintenance of Bcr/Abl-Positive Leukaemia. EMBO Mol. Med. 2010, 2, 98–110.
- Gallipoli, P.; Cook, A.; Rhodes, S.; Hopcroft, L.; Wheadon, H.; Whetton, A.D.; Jørgensen, H.G.; Bhatia, R.; Holyoake, T.L. JAK2/STAT5 Inhibition by Nilotinib with Ruxolitinib Contributes to the Elimination of CML CD34 + Cells in Vitro and in Vivo. Blood 2014, 124, 1492–1501.
- Chai, S.K.; Nichols, G.L.; Rothman, P. Constitutive activation of JAKs and STATs in BCR-Abl-expressing cell lines and peripheral blood cells derived from leukemic patients. J. Immunol. 2021, 159, 4720–4728.
- Fetisov, T.I.; Lesovaya, E.A.; Yakubovskaya, M.G.; Kirsanov, K.I.; Belitsky, G.A. Alterations in WNT Signaling in Leukemias. Biochemistry 2018, 83, 1448–1458.
- Soares-Lima, S.C.; Pombo-de-Oliveira, M.S.; Carneiro, F.R.G. The Multiple Ways Wnt Signaling Contributes to Acute Leukemia Pathogenesis. J. Leukoc. Biol. 2020, 108, 1081–1099.
- Janssens, V.; Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001, 353, 417–439.
- Gregor, T.; Bosakova, M.K.; Nita, A.; Abraham, S.P.; Fafilek, B.; Cernohorsky, N.H.; Rynes, J.; Foldynova-Trantirkova, S.; Zackova, D.; Mayer, J.; et al. Elucidation of Protein Interactions Necessary for the Maintenance of the BCR–ABL Signaling Complex. Cell. Mol. Life Sci. 2020, 77, 3885–3903.
- Bueno-da-Silva, A.E.B.; Brumatti, G.; Russo, F.O.; Green, D.R.; Amarante-Mendes, G.P. Bcr-Abl-Mediated Resistance to Apoptosis Is Independent of Constant Tyrosine-Kinase Activity. Cell Death Differ. 2003, 10, 592–598.
- Raitano, A.B.; Whang, Y.E.; Sawyers, C.L. Signal Transduction by Wild-Type and Leukemogenic Abl Proteins. Biochim. Biophys. Acta 1997, 1333, F201–F216.
- Sattler, M.; Salgia, R. Activation of Hematopoietic Growth Factor Signal Transduction Pathways by the Human Oncogene BCR/ABL. Cytokine Growth Factor Rev. 1997, 8, 63–79.
- Zou, X.; Calame, K. Signaling Pathways Activated by Oncogenic Forms of Abl Tyrosine Kinase. J. Biol. Chem. 1999, 274, 18141–18144.
- Dormer, P.; Lau, B.; Wilmanns, W. Kinetics of bone marrow cell production in human acute and chronic myeloid leukemias. Leuk. Res. 1980, 4, 231–237.
- Cortez, D.; Kadlec, L.; Pendergast, A.M. Structural and Signaling Requirements for BCR-ABL-Mediated Transformation and Inhibition of Apoptosis. Mol. Cell. Biol. 1995, 15, 5531–5541.
- Bedi, A.; Zehnbauer, B.A.; Barber, J.P.; Sharkis, S.J.; Jones, R.J. Inhibition of apoptosis by BCR-ABL in chronic myeloid leukemia. Blood 1994, 83, 2038–2044.
- Mcgahon, A.J.; Brown, D.G.; Martin, S.J.; Amarante-Mendes, G.P.; Cotter, T.G.; Cohen, G.M.; Green, D.R. Downregulation of Bcr-Abl in K562 Cells Restores Susceptibility to Apoptosis: Characterization of the Apoptotic Death. Cell Death Differ. 1997, 4, 95–104.
- Amarante-Mendes, G.P.; Finucane, D.M.; Martin, S.J.; Cotter, T.G.; Salvesen, G.S.; Green, D.R. Anti-Apoptotic Oncogenes Prevent Caspase-Dependent and Independent Commitment for Cell Death. Cell Death Differ. 1998, 5, 298–306.
- Fernandez-Luna, J.L. Bcr-Abl and Inhibition of Apoptosis in Chronic Myelogenous Leukemia Cells. Apoptosis 2000, 5, 315–318.
- Brumatti, G.; Weinlich, R.; Chehab, C.F.; Yon, M.; Amarante-Mendes, G.P. Comparison of the Anti-Apoptotic Effects of Bcr-Abl, Bcl-2 and Bcl-XL Following Diverse Apoptogenic Stimuli. FEBS Lett. 2003, 541, 57–63.
- Ghaffari, S.; Jagani, Z.; Kitidis, C.; Lodish, H.F.; Khosravi-Far, R. Cytokines and BCR-ABL Mediate Suppression of TRAIL-Induced Apoptosis through Inhibition of Forkhead FOXO3a Transcription Factor. Proc. Natl. Acad. Sci. USA 2003, 100, 6523–6528.
- Lugo, T.G.; Pendergast, A.M.; Muller, A.J.; Witte, O.N. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science 1990, 247, 1079–1082.
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 Recommendations for Treating Chronic Myeloid Leukemia. Leukemia 2020, 34, 966–984.
- Hehlmann, R. The New Eln Recommendations for Treating Cml. J. Clin. Med. 2020, 9, 3671.
- Khorashad, J.S.; Kelley, T.W.; Szankasi, P.; Mason, C.C.; Soverini, S.; Adrian, L.T.; Eide, C.A.; Zabriskie, M.S.; Lange, T.; Estrada, J.C.; et al. BCR-ABL1 Compound Mutations in Tyrosine Kinase Inhibitor-Resistant CML: Frequency and Clonal Relationships. Blood 2013, 121, 489–498.
- Soverini, S.; Branford, S.; Nicolini, F.E.; Talpaz, M.; Deininger, M.W.N.; Martinelli, G.; Müller, M.C.; Radich, J.P.; Shah, N.P. Implications of BCR-ABL1 Kinase Domain-Mediated Resistance in Chronic Myeloid Leukemia. Leuk. Res. 2014, 38, 10–20.
- Eck, M.J.; Manley, P.W. The Interplay of Structural Information and Functional Studies in Kinase Drug Design: Insights from BCR-Abl. Curr. Opin. Cell Biol. 2009, 21, 288–295.