 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jonathan Cortés-Martín | + 2507 word(s) | 2507 | 2020-09-03 05:49:54 | | | |
| 2 | Jonathan Cortés-Martín | -463 word(s) | 2044 | 2020-09-04 09:34:09 | | | | |
| 3 | Jonathan Cortés-Martín | -463 word(s) | 2044 | 2020-09-04 09:37:50 | | | | |
| 4 | Nicole Yin | Meta information modification | 2044 | 2020-09-09 05:04:37 | | |
Video Upload Options
Hajdu–Cheney syndrome (HCS) is a rare genetic disease that causes acroosteolysis and generalized osteoporosis, accompanied by a series of developmental skeletal disorders and multiple clinical and radiological manifestations. It has an autosomal dominant inheritance, although there are several sporadic non-hereditary cases.
1. Introduction
Hajdu–Cheney syndrome (HCS) is a rare genetic disease of the connective tissue that belongs to the osteolysis syndromes group. It is registered in the OMIM (Mendelian Inheritance in Man) project database with reference #102500 and in ORPHANET under the reference ORPHA955. It is also known as acro-dento-osteo dysplasia, acroosteolysis with osteoporosis and changes in the skull and mandible, arthrodentoosteodysplasia, and serpentine fibula-polycystic kidney syndrome. The prevalence of this disease is less than one person in one million (<1/1,000,000) and it is caused by a heterozygotic mutation of gene NOTCH2 located on chromosome 1p13-p11. HCS follows an autosomal-dominant inheritance pattern, although descriptions of cases with sporadic mutations can be found.
The disease was first described in 1948 by N. Hajdu and later completed by D. Cheney in 1965. Since then, around 50 cases of patients with HCS have been reported and, in general, all patients show a case of osteolysis of the distal phalanges and generalized osteoporosis, accompanied by other disorders, such as craniofacial and skeletal dysmorphia, developmental skeletal disorders, premature loss of teeth, and a short stature.
Due to the variability in the expression of NOTCH2, patients can be found with phenotypic diferences between them. Furthermore, this disease presents a wide and specific clinical spectrum that is rare to encounter in full in a single patient. Therefore, reports are found of cases diagnosed with HCS presenting variable clinical manifestations that worsen over time due to their age-dependent progression, with changes from early infancy to late adulthood.
2. Epidemiology
Hajdu–Cheney syndrome has a prevalence of less than 1 in 1,000,000 live births . Since 1948, approximately 50 cases have been described worldwide. It is a genetic disease with autosomal-dominant inheritance, although sporadic cases have been reported.
3. Etiology
As we have previously stated, HCS is a genetic disease caused by a heterozygotic mutation of NOTCH2.The NOTCH signaling pathway is constituted by a series of linked occurrences that are intimately related to skeletal development and homeostasis; therefore, alterations of this pathway cause disorders in both processes. NOTCH receptors are transmembrane proteins that have three major parts: an extracellular domain that consists of multiple EGF (epidermal growth factor)-like repeats, another intermembrane domain and an intracellular one that consists of multiple ankyrin repeats, nuclear localization signals, and a proline-, glutamic acid-, serine-, and threonine-rich domain, known as the PEST domain, whose function is the recycling of proteins. NOTCH has four receptors (NOTCH 1, 2, 3, and 4) and five ligands (JAG1, JAG2, and DLL 1, 2, and 4).
The NOTCH signaling pathway activates when a cell’s ligand adheres to the receptor of the cell, provoking the separation of the intracellular domain, which travels to the nucleus of the cell where it begins to complete its function.
In HCS, there is a truncation in exon 34 of NOTCH2, which causes a protein product to be missing the PEST domain, leading to an elevated level of NOTCH signaling activity in multiple tissues, therefore altering the usual process. This has a noticeable impact on skeletal development and homeostasis, leading to the disease.
Several disorders have been associated with NOTCH mutations along with HCS:
-CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and
leukoencephalopathy): mutations of EGF-like repeats of NOTCH3.
-Bicuspid aortic valve: protein-truncating mutations of NOTCH1.
-LAL-T: protein-truncating mutations of the PEST domain of NOTCH1.
- Alagille syndrome: mutation of the splice acceptor of exon 33 of NOTCH2.
4. Pathophysiology
Once a mutation has occurred, normal skeletal development is afected, causing a series of skeletal anomalies. There is a bone density déficit that leads to generalized skeletal dysplasia. Osteoporosis is one of the most characteristic signs of HCS, along with acroosteolysis of distal phalanges, both of which are caused by a series of local mechanisms that increase osteoclastic activity and impact bone formation negatively. Congenital defects in ossification can be found in fetal cartilage, causing peripheral dysostosis that worsens the acroosteolysis.
Understanding these processes helps us to comprehend some of the clinical manifestations that are seen in the wide spectrum of clinical presentations this syndrome provides: fractures in long bones due to bone demineralization, frequent respiratory infections caused by thoracic deformities and ventilatory restriction, basilar invagination and its neurological alterations, and short stature due to vertebral collapse. These are some of the clinical complications that arise from these processes that, when considered alongside the age-dependent progression of HCS, make this syndrome so complex.
5. Clinical Manifestations
This syndrome has three major characteristics regarding symptoms: a variable phenotype, a wide spectrum of clinical presentation, and an age-dependent progression. Following these three parameters, some patients are diagnosed with HCS with di_erent clinical presentations and phenotypic diferences person to person, and these manifestations tend to evolve over time. The following clinical manifestations are the most representative of HCS:
-Cranial alterations: bathrocephaly, presence of multiple wormian bones, delayed suture closure, thickened dome of the skull, absent frontal sinuses, elongated sella turcica, small jaw, basilar invagination, dolichocephaly, and occipital prominence.
-Facial alterations: coarse and dysmorphic facies, elongated philtrum, micrognathia, low-set ears,telecanthus, sinofridia, bushy eyebrows, long eyelashes, wide nose, high arched palate, premature denture loss, jaw malocclusion, hirsutism, and hypertelorism.
-Musculoskeletal alterations: short stature, short neck, fractures of long bones, joint laxity, biconcave vertebrae, kyphoscoliosis, cervical instability, vertebral collapse, genu valgum, serpentine fibula, acroosteolysis, pseudoclubbing, short fingers, Hippocratic fingers, progressive distal bone resorption, bone demineralization, osteopenia, and osteoporosis.
-Cardiovascular alterations: congenital heart disease, patent arterial duct, and sept defects.
-Digestive alterations: intestinal malrotation.
-Neurological alterations: hydrocephalus and lateral meningocele.
-Renal alterations: hypospadias, cryptorchidism, renal cysts, and kidney failure.
-Respiratory alterations: thoracic deformities, ventilatory restriction, and recurrent infections.
-Other alterations: delayed motor development, hearing loss, changes of the voice, deep voice, short nails, plantar ulcers, and hernias.
The most relevant clinical manifestations are shown in figures 1, 2,3 and 4.
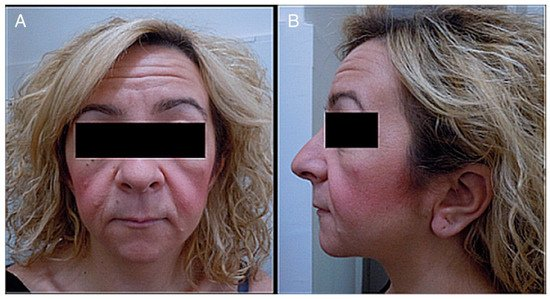
Figure 1. (A) Photograph of a patient’s face. (B) Lateral photograph of a patient’s head and face. The following are noted: small face, telecanthus and downslated palpebral fissures, micrognathia, small mouth, thin lips, long philtrum and full cheeks, low-positioned ears with a crease in the lobules, short neck, and coarse hair.
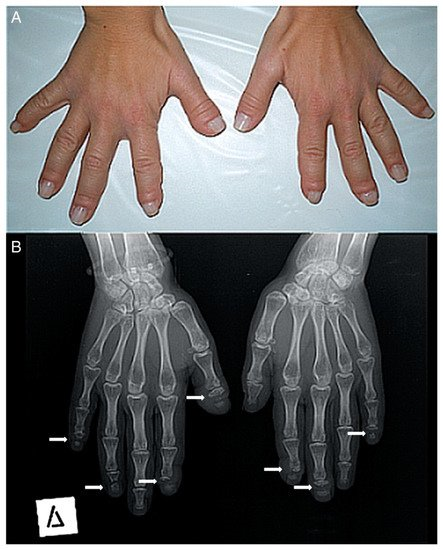
Figure 2. (A) Photograph of a patient’s hands. Many of her fingers are thick (predominately the right thumb) with pseudo-clubbing. (B) Anteroposterior radiograph of a patient’s hands. Osteolysis of the distal phalanges is found in most of the fingers (only the left thumb, the left fourth, and the right third fingers have a normal appearance). In all lesions, osteolysis has a transverse pattern across the width of the terminal phalanx (white arrows).
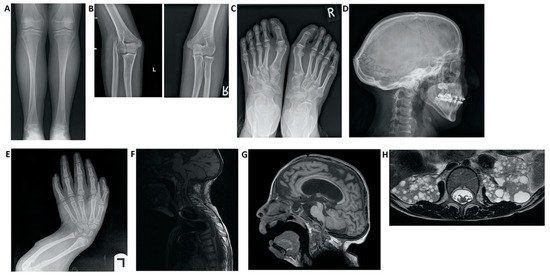
Figure 3. Imaging findings in patients with Hajdu–Cheney syndrome. (A) Radiograph of the frontal view of the tibia and fibula. Note the elongated and medially deviated fibula, superposing the tibia bone, referred to as a “serpentine fibula.” (B) Radiographs of the lateral view of the left arm showing dislocation of the radial head. (C) Radiograph of the frontal view of the feet showing hallux valgus, crowded metatarsal bones, short foot distal digits, and acroosteolysis signs at distal phalanges of the first digit. (D) Radiograph of the lateral view of the skull, demonstrating batrocephaly, frontal sinuses hypoplasia, and multiple occipital wormian bones. (E) Radiograph of the frontal view of the hands, showing crowded metacarpal bones, short hand distal digits, and acroosteolysis signs at the distal phalanges. (E) Radiograph of the lateral view of the arm, showing shortening of the distal fingers and long bones of the arm, and a curved radius and ulna. (F) Spinal MRI showing deformations of the cervical spine, as well as a significant syrinx of the cord. (G) Brain MRI. Note the ventricular enlargement with a VP (ventriculo-peritoneal) shunt, frontal hypoplasia of the sinuses, and a small foramen magnum. (H) Abdominal MRI showing multiple bilateral kidney cysts.
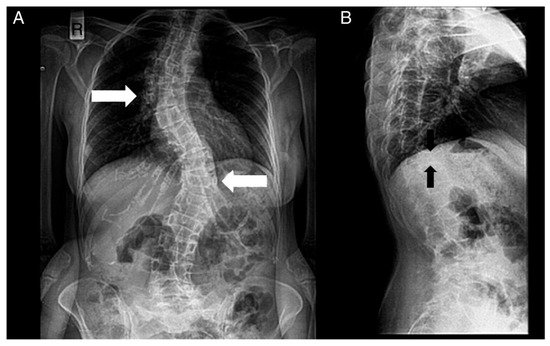
Figure 4. Radiographs of a patient’s spine. (A) Anteroposterior view. (B) Lateral view. The following are noted: scoliosis (double major—with a right thoracic and a left thoracolumbar curve—white noted: scoliosis (double major—with a right thoracic and a left thoracolumbar curve—white arrows), and biconcave deformities of the upper and lower endplate (fishbone deformity—black arrows) of many vertebrae and decreased bone density.
The most frequent clinical complications in this syndrome are basilar invagination, and consequently, brain damage, hydrocephalus, syringomyelia, vertebral collapse due to compression, and ventilatory restriction caused by a thoracic deformity.
There is a subgroup of patients within this syndrome that present two distinctive signs: serpentine fibula and polycystic kidneys. Initially, it was thought that patients with these signs belonged to a separate disease, di_erent from HCS, but genetic studies demonstrated both originate from the same mutated gene. The variable expression of NOTCH2 justifies the frequent association of serpentine fibula and polycystic kidneys as nothing but another manifestation of HCS and not an independent syndrome.
Based on the age-dependent progression of this syndrome, it has been shown that after monitoring several cases over time, the phenotypes and symptoms gradually worsen. There are a series of stages of the disease according to age, allowing for seven generational divisions:
-birth (<1 year old)
-early childhood (ages 1–5)
-childhood (ages 6–12)
-adolescence (ages 13–19)
-early adulthood (ages 20–33)
-middle adulthood (ages 36–65)
-late adulthood (65+).
We highlight the importance of presenting an updated report of the variability of manifestations and the changing phenotype of this disease.
6. Diagnosis
The diagnosis of HCS is suspected via the observation of physical appearance and radiological findings, but the final diagnosis is reached via genetic sequencing of exon 34 of NOTCH2.
Brennan and Pauli created a diagnostic tool that establishes the inclusion criteria for this syndrome.
It is worth highlighting the need for establishing a diferential diagnosis with other disorders and syndromes that share clinical manifestations and could generate diagnostic uncertainty.
Regarding its osteolytic nature, HCS can be compared to some of the disorders that belong to the group of osteolysis syndromes, such as Torg, François, Whyte–Hemingway,Winchester, and a new syndrome known as Talo-patello-scaphoid osteolysis, synovitis, and short fourth metacarpals.One of the main characteristics of the disease to be analyzed is acroosteolysis but we can also find other disorders that present as signs, such as scleroderma, sarcoidosis, neuropathic disorders, and rheumatoid syndromes. Progeria and pycnodysostosis are another type of disorder that cause congenital acroosteolysis. Including Paget’s disease or other osteoporosis syndromes in the diferential diagnosis is also of interest due to their osteoporotic nature. There are studies on the diferential diagnosis between HCS and lateral meningocele considering phenotypic similarities and with Alagille syndrome because of their genetic links. Other syndromes that share basilar invagination amongst their clinical manifestations are osteogenesis imperfecta, congenital osteochondrodysplasia and spondyloepiphyseal dysplasia, and could also be considered when creating the diferential diagnosis for HCS.
7. Treatment
There is no definitive or efective pharmacological treatment for HCS at present, and although certain trials with bisphosphonates have been developed, there is insufcient evidence of their efectiveness. Surgical intervention as a method to avoid complications has proven to be efective in certain cases. The current treatment for HCS is based on the management of complications and underlying problems in order to improve the patient’s quality of life and life expectancy. Certain studies consider the manipulation of gamma-secretase inhibitors as a possible way to prevent this disorder.
8. Prognosis
HCS is classified as a rare genetic disease but there are no studies that offer a global perspective on the prognosis and quality of life of afected patients. The severity of the disease depends on the afected organs, clinical complications, and the degenerative evolution of each patient. The generalized osteoporosis and the development of acroosteolysis will cause fractures, difculty with walking, and dependency for everyday life activities.
The prognosis worsens when complications such as basilar invagination exist, causing neurologic alterations, or thoracic deformities that cause ventilatory restriction. Due to the low prevalence and the lack of qualitative information about this syndrome, it is di_cult to know the burden of disease and the years of healthy life lost. Researchers should discuss the results and how they can be interpreted given previous studies and the working hypotheses. The findings and their implications should be discussed in the broadest context possible. Future research directions may also be highlighted.

