Hajdu–Cheney syndrome (HCS) is a rare genetic disease that causes acroosteolysis and generalized osteoporosis, accompanied by a series of developmental skeletal disorders and multiple clinical and radiological manifestations. It has an autosomal dominant inheritance, although there are several sporadic non-hereditary cases.El síndrome de Hajdu-Cheney (HCS) es una enfermedad genética rara que causa acroosteólisis y osteoporosis generalizada, acompañada de una serie de trastornos esqueléticos del desarrollo y múltiples manifestaciones clínicas y radiológicas. Tiene una herencia autosómica dominante, aunque hay varios casos esporádicos no hereditarios.
- Enfermedades raras
- Hajdu-Cheney
- NOTCH2
- Tejido conectivo
1. Introduction1. Introducción
Hajdu–Cheney syndrome (HCS) is a rare genetic disease of the connective tissue that belongs to the osteolysis syndromes group. It is registered in the OMIM (Mendelian Inheritance in Man) project database with reference #102500 and in ORPHANET under the reference ORPHA955. It is also known as acro-dento-osteo dysplasia, acroosteolysis with osteoporosis and changes in the skull and mandible, arthrodentoosteodysplasia, and serpentine fibula-polycystic kidney syndrome. The prevalence of this disease is less than one person in one million (<1/1,000,000) and it is caused by a heterozygotic mutation of gene NOTCH2 located on chromosome 1p13-p11. HCS follows an autosomal-dominant inheritance pattern, although descriptions of cases with sporadic mutations can be found.El síndrome de Hajdu-Cheney (HCS) es una enfermedad genética rara del tejido conectivo que pertenece al grupo de los síndromes de osteólisis. Está registrado en la base de datos del proyecto OMIM (Herencia mendeliana en el hombre) con referencia # 102500 y en ORPHANET con la referencia ORPHA955. También se conoce como displasia acro-dento-osteo, acroosteólisis con osteoporosis y cambios en el cráneo y mandíbula, artrodentoosteodisplasia y síndrome de peroné serpentino-riñón poliquístico. La prevalencia de esta enfermedad es menos de una persona en un millón (<1 / 1.000.000) y está causada por una mutación heterocigótica del gen NOTCH 2 localizado en el cromosoma 1p13-p11.El HCS sigue un patrón de herencia autosómico dominante, aunque se pueden encontrar descripciones de casos con mutaciones esporádicas.
The disease was first described in 1948 by N. Hajdu and later completed by D. Cheney in 1965. Since then, around 50 cases of patients with HCS have been reported and, in general, all patients show a case of osteolysis of the distal phalanges and generalized osteoporosis, accompanied by other disorders, such as craniofacial and skeletal dysmorphia, developmental skeletal disorders, premature loss of teeth, and a short stature.La enfermedad fue descrita por primera vez en 1948 por N. Hajdu [6] y posteriormente completada por D. Cheney en 1965. Desde entonces, se han reportado alrededor de 50 casos de pacientes con HCS y, en general, todos los pacientes mostrar un caso de osteólisis de las falanges distales y osteoporosis generalizada, acompañada de otros trastornos, como dismorfia craneofacial y esquelética, desarrollo esquelético trastornos, pérdida prematura de dientes y baja estatura.
Due to the variability in the expression of NOTCH2, patients can be found with phenotypic diferences between them. Furthermore, this disease presents a wide and specific clinical spectrum that is rare to encounter in full in a single patient. Therefore, reports are found of cases diagnosed with HCS presenting variable clinical manifestations that worsen over time due to their age-dependent progression, with changes from early infancy to late adulthood.Debido a la variabilidad en la expresión de NOTCH2 , se pueden encontrar pacientes con diferencias fenotípicas entre ellos. Además, esta enfermedad presenta un espectro clínico amplio y específico que es raro encontrar en su totalidad en un solo paciente. Por tanto, se encuentran reportes de casos diagnosticados de HCS que presentan manifestaciones clínicas variables que empeoran con el tiempo debido a su progresión dependiente de la edad, con cambios desde la primera infancia hasta la edad adulta tardía.
2. Epidemiology 2. Epidemiología
Hajdu–Cheney syndrome has a prevalence of less than 1 in 1,000,000 live births . Since 1948, approximately 50 cases have been described worldwide. It is a genetic disease with autosomal-dominant inheritance, although sporadic cases have been reported.El síndrome de Hajdu-Cheney tiene una prevalencia de menos de 1 de cada 1.000.000 de nacidos vivos. Desde 1948, se han descrito aproximadamente 50 casos en todo el mundo. Es una enfermedad genética con herencia autosómica dominante, aunque se han informado casos esporádicos.
3. Etiology3. Etiología
As we have previously stated, HCS is a genetic disease caused by a heterozygotic mutation of NOTCH2.The NOTCH signaling pathway is constituted by a series of linked occurrences that are intimately related to skeletal development and homeostasis; therefore, alterations of this pathway cause disorders in both processes. NOTCH receptors are transmembrane proteins that have three major parts: an extracellular domain that consists of multiple EGF (epidermal growth factor)-like repeats, another intermembrane domain and an intracellular one that consists of multiple ankyrin repeats, nuclear localization signals, and a proline-, glutamic acid-, serine-, and threonine-rich domain, known as the PEST domain, whose function is the recycling of proteins. NOTCH has four receptors (NOTCH 1, 2, 3, and 4) and five ligands (JAG1, JAG2, and DLL 1, 2, and 4).Como hemos dicho anteriormente, el HCS es una enfermedad genética causada por una mutación heterocigótica de NOTCH2 . La vía de señalización NOTCH está constituida por una serie de sucesos vinculados que están íntimamente relacionados con el desarrollo esquelético y la homeostasis; por tanto, las alteraciones de esta vía provocan alteraciones en ambos procesos.
The NOTCH signaling pathway activates when a cell’s ligand adheres to the receptor of the cell, provoking the separation of the intracellular domain, which travels to the nucleus of the cell where it begins to complete its function.Los receptores NOTCH son proteínas transmembrana que tienen tres partes principales: un dominio extracelular que consta de múltiples repeticiones similares al EGF (factor de crecimiento epidérmico), otro dominio intermembrana y uno intracelular que consta de múltiples repeticiones de anquirina, señales de localización nuclear, y un dominio rico en prolina, ácido glutámico, serina y treonina, conocido como dominio PEST, cuya función es el reciclaje de proteínas. NOTCH tiene cuatro receptores ( NOTCH 1, 2, 3 y 4) y cinco ligandos (JAG1, JAG2 y DLL 1, 2 y 4).
In HCS, there is a truncation in exon 34 of NOTCH2, which causes a protein product to be missing the PEST domain, leading to an elevated level of NOTCH signaling activity in multiple tissues, therefore altering the usual process. This has a noticeable impact on skeletal development and homeostasis, leading to the disease.La vía de señalización NOTCH se activa cuando el ligando de una célula se adhiere al receptor de la célula, provocando la separación del dominio intracelular, que viaja al núcleo de la célula donde comienza a completar su función.
Several disorders have been associated with NOTCH mutations along with HCS:En HCS, hay un truncamiento en el exón 34 de NOTCH2, lo que hace que un producto proteico no tenga el dominio PEST, lo que lleva a un nivel elevado de actividad de señalización de NOTCH en múltiples tejidos, alterando así el proceso habitual. Esto tiene un impacto notable en el desarrollo esquelético y la homeostasis, lo que conduce a la enfermedad.
-CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts andSe han asociado varios trastornos con mutaciones NOTCH junto con HCS:
leukoencephalopathy): mutations of EGF-like repeats of NOTCH3.
- CADASIL (arteriopatía cerebral autosómica dominante con infartos subcorticales y leucoencefalopatía): mutaciones de repeticiones tipo EGF de NOTCH3 .
- Válvula aórtica bicúspide: mutaciones truncadoras de proteínas de NOTCH1 .
- LAL-T: mutaciones que truncan proteínas del dominio PEST de NOTCH1 .
- Síndrome de Alagille: mutación del aceptor de empalme del exón 33 de NOTCH2 .
-Bicuspid aortic valve: protein-truncating mutations of NOTCH1.
4. Fisiopatología
-LAL-T: protein-truncating mutations of the PEST domain of NOTCH1.Una vez que se ha producido una mutación, el desarrollo esquelético normal se ve afectado, lo que provoca una serie de anomalías esqueléticas. Existe un déficit de densidad ósea que conduce a una displasia esquelética generalizada. La osteoporosis es uno de los signos más característicos del HCS, junto con la acroosteólisis de las falanges distales, ambas causadas por una serie de mecanismos locales que aumentan la actividad osteoclástica e impactan negativamente en la formación de hueso. Se pueden encontrar defectos congénitos en la osificación en el cartílago fetal, que provocan disostosis periférica que empeora la acroosteólisis.
- Alagille syndrome: mutation of the splice acceptor of exon 33 of NOTCH2.La comprensión de estos procesos nos ayuda a comprender algunas de las manifestaciones clínicas que se observan en el amplio espectro de presentaciones clínicas que proporciona este síndrome: fracturas en huesos largos por desmineralización ósea, infecciones respiratorias frecuentes causadas por deformidades torácicas y restricción ventilatoria, invaginación basilar y sus alteraciones neurológicas, y baja estatura por colapso vertebral. Estas son algunas de las complicaciones clínicas que surgen de estos procesos que, cuando se consideran junto con la progresión dependiente de la edad del HCS, hacen que este síndrome sea tan complejo.
4. Pathophysiology5. Manifestaciones clínicas
Once a mutation has occurred, normal skeletal development is afected, causing a series of skeletal anomalies. There is a bone density déficit that leads to generalized skeletal dysplasia. Osteoporosis is one of the most characteristic signs of HCS, along with acroosteolysis of distal phalanges, both of which are caused by a series of local mechanisms that increase osteoclastic activity and impact bone formation negatively. Congenital defects in ossification can be found in fetal cartilage, causing peripheral dysostosis that worsens the acroosteolysis.Este síndrome tiene tres características principales con respecto a los síntomas: un fenotipo variable, un amplio espectro de presentación clínica y una progresión dependiente de la edad. Siguiendo estos tres parámetros, algunos pacientes son diagnosticados de HCS con diferentes presentaciones clínicas y diferencias fenotípicas de persona a persona, y estas manifestaciones tienden a evolucionar con el tiempo. Las siguientes manifestaciones clínicas son las más representativas de HCS:
Understanding these processes helps us to comprehend some of the clinical manifestations that are seen in the wide spectrum of clinical presentations this syndrome provides: fractures in long bones due to bone demineralization, frequent respiratory infections caused by thoracic deformities and ventilatory restriction, basilar invagination and its neurological alterations, and short stature due to vertebral collapse. These are some of the clinical complications that arise from these processes that, when considered alongside the age-dependent progression of HCS, make this syndrome so complex.
- Alteraciones craneales: batrocefalia, presencia de múltiples huesos wormianos, cierre de sutura retardado, domo engrosado del cráneo, ausencia de senos frontales, silla turca alargada, mandíbula pequeña, invaginación basilar, dolicocefalia y prominencia occipital.
- Alteraciones faciales: facies gruesa y dismórfica, filtrum alargado, micrognatia, orejas de implantación baja, telecanto, sinofridia, cejas tupidas, pestañas largas, nariz ancha, paladar alto arqueado, pérdida prematura de la dentadura, maloclusión mandibular, hirsutismo e hipertelorismo.
- Alteraciones musculoesqueléticas: talla baja, cuello corto, fracturas de huesos largos, laxitud articular, vértebras bicóncavas, cifoescoliosis, inestabilidad cervical, colapso vertebral, genu valgo, peroné serpentino, acroosteólisis, pseudoclubbing, dedos cortos, dedos hipocráticos, resorción ósea distal progresiva, hueso desmineralización, osteopenia y osteoporosis.
- Alteraciones cardiovasculares: cardiopatía congénita, conducto arterial permeable y defectos del tabique.
- Alteraciones digestivas: malrotación intestinal.
- Alteraciones neurológicas: hidrocefalia y meningocele lateral.
- Alteraciones renales: hipospadias, criptorquidia, quistes renales e insuficiencia renal.
- Alteraciones respiratorias: deformidades torácicas, restricción ventilatoria e infecciones recurrentes.
- Otras alteraciones: retraso en el desarrollo motor, hipoacusia, alteraciones de la voz, voz grave, uñas cortas, úlceras plantares y hernias.
5. Clinical Manifestations
Las manifestaciones clínicas más relevantes se muestran en las Figuras 1, 2, 3 y 4.
This syndrome has three major characteristics regarding symptoms: a variable phenotype, a wide spectrum of clinical presentation, and an age-dependent progression. Following these three parameters, some patients are diagnosed with HCS with di_erent clinical presentations and phenotypic diferences person to person, and these manifestations tend to evolve over time. The following clinical manifestations are the most representative of HCS: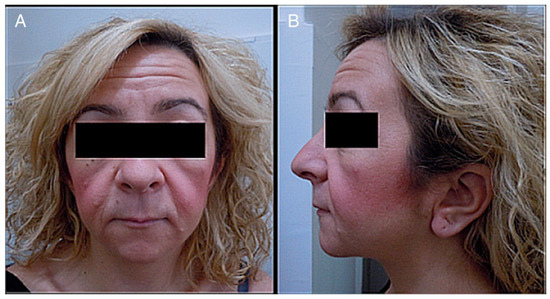
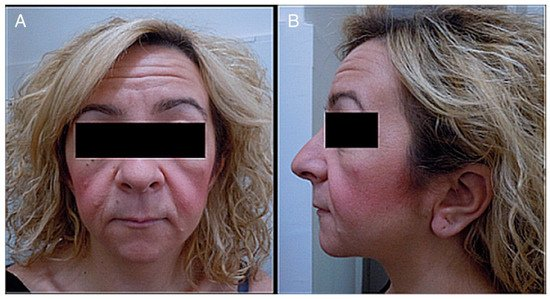
-Cranial alterations: bathrocephaly, presence of multiple wormian bones, delayed suture closure, thickened dome of the skull, absent frontal sinuses, elongated sella turcica, small jaw, basilar invagination, dolichocephaly, and occipital prominence.Figura 1. ( A ) Fotografía del rostro de un paciente. ( B ) Fotografía lateral de la cabeza y el rostro de un paciente. Se observa lo siguiente: cara pequeña, telecanto y fisuras palpebrales descendentes, micrognatia, boca pequeña, labios finos, surco nasolabial largo y mejillas llenas, orejas bajas con pliegue en los lóbulos, cuello corto y pelo áspero.
-Facial alterations: coarse and dysmorphic facies, elongated philtrum, micrognathia, low-set ears,telecanthus, sinofridia, bushy eyebrows, long eyelashes, wide nose, high arched palate, premature denture loss, jaw malocclusion, hirsutism, and hypertelorism.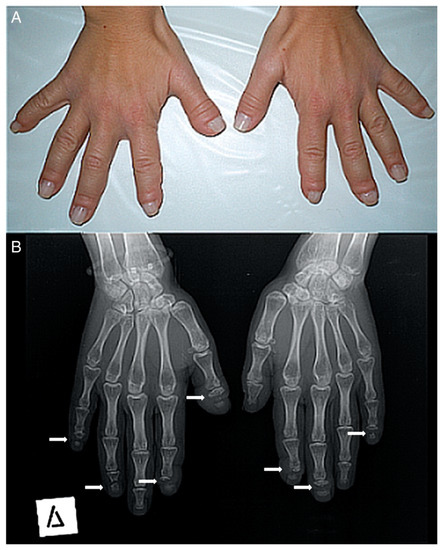
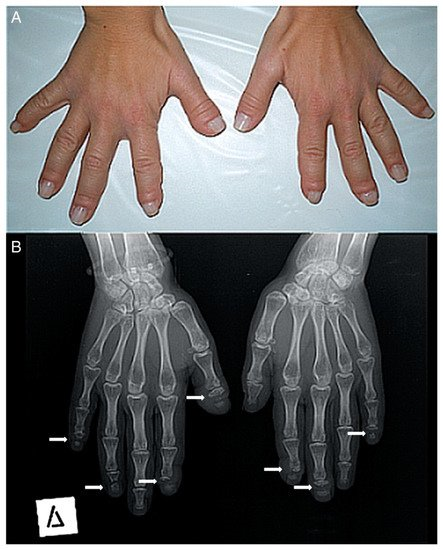
-Musculoskeletal alterations: short stature, short neck, fractures of long bones, joint laxity, biconcave vertebrae, kyphoscoliosis, cervical instability, vertebral collapse, genu valgum, serpentine fibula, acroosteolysis, pseudoclubbing, short fingers, Hippocratic fingers, progressive distal bone resorption, bone demineralization, osteopenia, and osteoporosis.Figura 2. ( A ) Fotografía de las manos de un paciente. Muchos de sus dedos son gruesos (predominantemente el pulgar derecho) con pseudo-discotecas. ( B ) Radiografía anteroposterior de las manos de un paciente. La osteólisis de las falanges distales se encuentra en la mayoría de los dedos (solo el pulgar izquierdo, el cuarto izquierdo y el tercer dedo derecho tienen un aspecto normal). En todas las lesiones, la osteólisis tiene un patrón transversal a lo ancho de la falange terminal (flechas blancas).
-Cardiovascular alterations: congenital heart disease, patent arterial duct, and sept defects.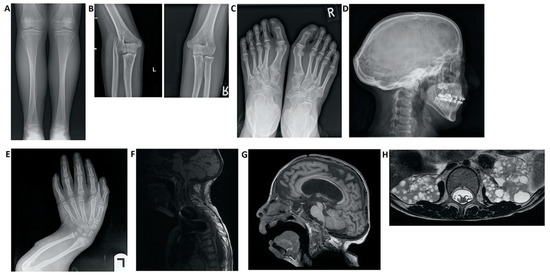
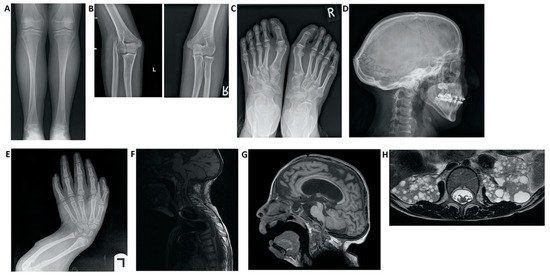
-Digestive alterations: intestinal malrotation.Figura 3. Hallazgos de imagen en pacientes con síndrome de Hajdu-Cheney. ( A ) Radiografía de la vista frontal de la tibia y el peroné. Observe el peroné alargado y medialmente desviado, que se superpone al hueso de la tibia, denominado "peroné serpentino". ( B ) Radiografías de la vista lateral del brazo izquierdo que muestran luxación de la cabeza radial. ( C ) Radiografía de la vista frontal de los pies que muestra hallux valgus, huesos metatarsianos apiñados, dedos distales del pie corto y signos de acroosteolisis en las falanges distales del primer dedo. ( D ) Radiografía de la vista lateral del cráneo, que demuestra batrocefalia, hipoplasia de los senos frontales y múltiples huesos de gusano occipital. ( E) Radiografía de la vista frontal de las manos, que muestra apiñamiento de los huesos metacarpianos, dedos cortos distales de la mano y signos de acroosteolisis en las falanges distales. ( E ) Radiografía de la vista lateral del brazo, que muestra acortamiento de los dedos distales y huesos largos del brazo, y un radio y cúbito curvados. ( F ) RM de columna que muestra deformaciones de la columna cervical, así como una siringe importante de la médula. ( G ) Resonancia magnética cerebral. Nótese el agrandamiento ventricular con una derivación VP (ventrículo-peritoneal), hipoplasia frontal de los senos nasales y un pequeño foramen magnum. ( H ) RM abdominal que muestra múltiples quistes renales bilaterales.
-Neurological alterations: hydrocephalus and lateral meningocele.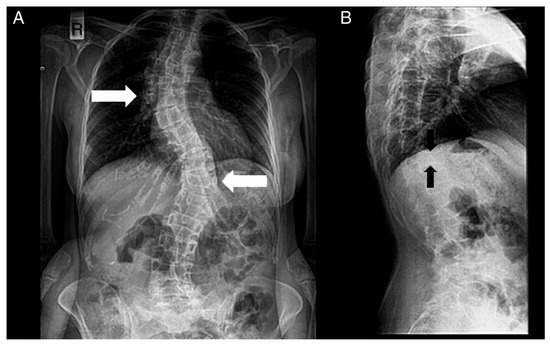
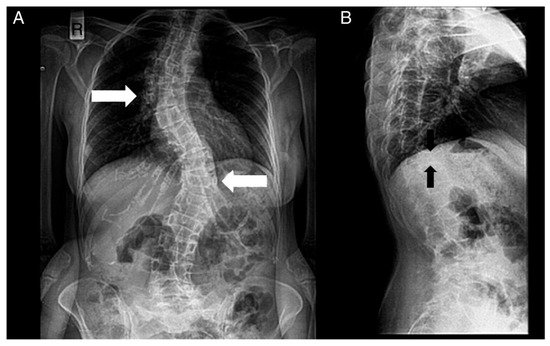
-Renal alterations: hypospadias, cryptorchidism, renal cysts, and kidney failure.Figura 4. Radiografías de la columna de un paciente. ( A ) Vista anteroposterior. ( B ) Vista lateral. Se notan los siguientes: escoliosis (doble mayor — con una curva torácica derecha y toracolumbar izquierda — flechas blancas) y deformidades bicóncavas de la placa terminal superior e inferior (deformidad en espina de pescado — flechas negras) de muchas vértebras y disminución de la densidad ósea.
-Respiratory alterations: thoracic deformities, ventilatory restriction, and recurrent infections.Las complicaciones clínicas más frecuentes de este síndrome son la invaginación basilar y, en consecuencia, el daño cerebral, la hidrocefalia, la siringomielia, el colapso vertebral debido a la compresión y la restricción ventilatoria causada por una deformidad torácica.
-Other alterations: delayed motor development, hearing loss, changes of the voice, deep voice, short nails, plantar ulcers, and hernias.Existe un subgrupo de pacientes dentro de este síndrome que presentan dos signos distintivos: peroné serpentino y riñones poliquísticos. Inicialmente, se pensaba que los pacientes con estos signos pertenecían a una enfermedad separada, diferente de HCS, pero los estudios genéticos demostraron que ambos se originaban en el mismo gen mutado. La expresión variable de NOTCH2 justifica la asociación frecuente de peroné serpentino y riñones poliquísticos como nada más que otra manifestación de HCS y no un síndrome independiente.
The most relevant clinical manifestations are shown in figures 1, 2,3 and 4.En base a la progresión de este síndrome dependiente de la edad, se ha demostrado que después de monitorear varios casos a lo largo del tiempo, los fenotipos y síntomas empeoran gradualmente. Hay una serie de estadios de la enfermedad según la edad, que permiten siete divisiones generacionales:
- nacimiento (<1 año)
- primera infancia (de 1 a 5 años)
- infancia (de 6 a 12 años)
- adolescencia (13 a 19 años)
- adultez temprana (20 a 33 años)
- edad adulta media (36 a 65 años)
- adultez tardía (65+).
Figure 1. (A) Photograph of a patient’s face. (B) Lateral photograph of a patient’s head and face. The following are noted: small face, telecanthus and downslated palpebral fissures, micrognathia, small mouth, thin lips, long philtrum and full cheeks, low-positioned ears with a crease in the lobules, short neck, and coarse hair.Destacamos la importancia de presentar un informe actualizado de la variabilidad de las manifestaciones y el fenotipo cambiante de esta enfermedad.
6. Diagnóstico
Figure 2. (A) Photograph of a patient’s hands. Many of her fingers are thick (predominately the right thumb) with pseudo-clubbing. (B) Anteroposterior radiograph of a patient’s hands. Osteolysis of the distal phalanges is found in most of the fingers (only the left thumb, the left fourth, and the right third fingers have a normal appearance). In all lesions, osteolysis has a transverse pattern across the width of the terminal phalanx (white arrows).El diagnóstico de HCS se sospecha mediante la observación de la apariencia física y los hallazgos radiológicos [29], pero el diagnóstico final se llega a través de la secuenciación genética del exón 34 de NOTCH2 .
Brennan y Pauli crearon una herramienta de diagnóstico que establece los criterios de inclusión para este síndrome.
Figure 3. Imaging findings in patients with Hajdu–Cheney syndrome. (A) Radiograph of the frontal view of the tibia and fibula. Note the elongated and medially deviated fibula, superposing the tibia bone, referred to as a “serpentine fibula.” (B) Radiographs of the lateral view of the left arm showing dislocation of the radial head. (C) Radiograph of the frontal view of the feet showing hallux valgus, crowded metatarsal bones, short foot distal digits, and acroosteolysis signs at distal phalanges of the first digit. (D) Radiograph of the lateral view of the skull, demonstrating batrocephaly, frontal sinuses hypoplasia, and multiple occipital wormian bones. (E) Radiograph of the frontal view of the hands, showing crowded metacarpal bones, short hand distal digits, and acroosteolysis signs at the distal phalanges. (E) Radiograph of the lateral view of the arm, showing shortening of the distal fingers and long bones of the arm, and a curved radius and ulna. (F) Spinal MRI showing deformations of the cervical spine, as well as a significant syrinx of the cord. (G) Brain MRI. Note the ventricular enlargement with a VP (ventriculo-peritoneal) shunt, frontal hypoplasia of the sinuses, and a small foramen magnum. (H) Abdominal MRI showing multiple bilateral kidney cysts.Cabe destacar la necesidad de establecer un diagnóstico diferencial con otros trastornos y síndromes que comparten manifestaciones clínicas y pueden generar incertidumbre diagnóstica.
En cuanto a su naturaleza osteolítica, el HCS se puede comparar con algunos de los trastornos que pertenecen al grupo de síndromes de osteólisis, como Torg, François, Whyte-Hemingway, Winchester, y un nuevo síndrome conocido como osteólisis Talo-patelo-escafoidea , sinovitis y cuartos metacarpianos cortos. Una de las principales características de la enfermedad a analizar es la acroosteólisis, que es un signo que también podemos encontrar en otros trastornos como la esclerodermia, sarcoidosis, trastornos neuropáticos y síndromes reumatoides. La progeria y la picnodisostosis son otro tipo de trastorno que causa acroosteolisis congénita. Incluir la enfermedad de Paget u otros síndromes de osteoporosis en el diagnóstico diferencial también es de interés por su naturaleza osteoporótica. Existen estudios sobre el diagnóstico diferencial entre HCS y meningocele lateral considerando similitudes fenotípicas y con el síndrome de Alagille debido a sus vínculos genéticos. Otros síndromes que comparten la invaginación basilar entre sus manifestaciones clínicas son la osteogénesis imperfecta, la osteocondrodisplasia congénita y la displasia espondiloepifisaria, y también podrían tenerse en cuenta al crear el diagnóstico diferencial de HCS.
Figure 4. Radiographs of a patient’s spine. (A) Anteroposterior view. (B) Lateral view. The following are noted: scoliosis (double major—with a right thoracic and a left thoracolumbar curve—white noted: scoliosis (double major—with a right thoracic and a left thoracolumbar curve—white arrows), and biconcave deformities of the upper and lower endplate (fishbone deformity—black arrows) of many vertebrae and decreased bone density.
7. Tratamiento
The most frequent clinical complications in this syndrome are basilar invagination, and consequently, brain damage, hydrocephalus, syringomyelia, vertebral collapse due to compression, and ventilatory restriction caused by a thoracic deformity.No existe un tratamiento farmacológico definitivo o eficaz para el HCS en la actualidad, y aunque se han desarrollado ciertos ensayos con bifosfonatos, no hay pruebas suficientes de su eficacia. La intervención quirúrgica como método para evitar complicaciones ha demostrado ser eficaz en determinados casos. El tratamiento actual para el HCS se basa en el manejo de complicaciones y problemas subyacentes con el fin de mejorar la calidad de vida y la esperanza de vida del paciente. Ciertos estudios consideran la manipulación de inhibidores de la gamma-secretasa como una posible forma de prevenir este trastorno.
There is a subgroup of patients within this syndrome that present two distinctive signs: serpentine fibula and polycystic kidneys. Initially, it was thought that patients with these signs belonged to a separate disease, di_erent from HCS, but genetic studies demonstrated both originate from the same mutated gene. The variable expression of NOTCH2 justifies the frequent association of serpentine fibula and polycystic kidneys as nothing but another manifestation of HCS and not an independent syndrome.
8. Pronóstico
Based on the age-dependent progression of this syndrome, it has been shown that after monitoring several cases over time, the phenotypes and symptoms gradually worsen. There are a series of stages of the disease according to age, allowing for seven generational divisions:El HCS está clasificado como una enfermedad genética rara, pero no existen estudios que ofrezcan una perspectiva global sobre el pronóstico y la calidad de vida de los pacientes afectados. La gravedad de la enfermedad depende de los órganos afectados, las complicaciones clínicas y la evolución degenerativa de cada paciente. La osteoporosis generalizada y el desarrollo de acroosteolisis provocarán fracturas, dificultad para caminar y dependencia para las actividades de la vida diaria.
-birth (<1 year old)
-early childhood (ages 1–5)
-childhood (ages 6–12)
-adolescence (ages 13–19)
-early adulthood (ages 20–33)
-middle adulthood (ages 36–65)
-late adulthood (65+).
We highlight the importance of presenting an updated report of the variability of manifestations and the changing phenotype of this disease.
6. Diagnosis
The diagnosis of HCS is suspected via the observation of physical appearance and radiological findings, but the final diagnosis is reached via genetic sequencing of exon 34 of NOTCH2.
Brennan and Pauli created a diagnostic tool that establishes the inclusion criteria for this syndrome.
It is worth highlighting the need for establishing a diferential diagnosis with other disorders and syndromes that share clinical manifestations and could generate diagnostic uncertainty.
Regarding its osteolytic nature, HCS can be compared to some of the disorders that belong to the group of osteolysis syndromes, such as Torg, François, Whyte–Hemingway,Winchester, and a new syndrome known as Talo-patello-scaphoid osteolysis, synovitis, and short fourth metacarpals.One of the main characteristics of the disease to be analyzed is acroosteolysis but we can also find other disorders that present as signs, such as scleroderma, sarcoidosis, neuropathic disorders, and rheumatoid syndromes. Progeria and pycnodysostosis are another type of disorder that cause congenital acroosteolysis. Including Paget’s disease or other osteoporosis syndromes in the diferential diagnosis is also of interest due to their osteoporotic nature. There are studies on the diferential diagnosis between HCS and lateral meningocele considering phenotypic similarities and with Alagille syndrome because of their genetic links. Other syndromes that share basilar invagination amongst their clinical manifestations are osteogenesis imperfecta, congenital osteochondrodysplasia and spondyloepiphyseal dysplasia, and could also be considered when creating the diferential diagnosis for HCS.
7. Treatment
There is no definitive or efective pharmacological treatment for HCS at present, and although certain trials with bisphosphonates have been developed, there is insufcient evidence of their efectiveness. Surgical intervention as a method to avoid complications has proven to be efective in certain cases. The current treatment for HCS is based on the management of complications and underlying problems in order to improve the patient’s quality of life and life expectancy. Certain studies consider the manipulation of gamma-secretase inhibitors as a possible way to prevent this disorder.
8. Prognosis
HCS is classified as a rare genetic disease but there are no studies that offer a global perspective on the prognosis and quality of life of afected patients. The severity of the disease depends on the afected organs, clinical complications, and the degenerative evolution of each patient. The generalized osteoporosis and the development of acroosteolysis will cause fractures, difculty with walking, and dependency for everyday life activities.
The prognosis worsens when complications such as basilar invagination exist, causing neurologic alterations, or thoracic deformities that cause ventilatory restriction. Due to the low prevalence and the lack of qualitative information about this syndrome, it is di_cult to know the burden of disease and the years of healthy life lost. Researchers should discuss the results and how they can be interpreted given previous studies and the working hypotheses. The findings and their implications should be discussed in the broadest context possible. Future research directions may also be highlighted.El pronóstico empeora cuando existen complicaciones como la invaginación basilar, que provocan alteraciones neurológicas o deformidades torácicas que provocan restricción ventilatoria. Debido a la baja prevalencia y la falta de información cualitativa sobre este síndrome, es difícil conocer la carga de enfermedad y los años de vida saludable perdidos. Los investigadores deben discutir los resultados y cómo se pueden interpretar los datos de los estudios previos y las hipótesis de trabajo. Los hallazgos y sus implicaciones deben discutirse en el contexto más amplio posible. También se pueden destacar las direcciones de investigación futuras.