1. Retinal Diseases Directly or Indirectly Associated with Vitamin A and Its Pathways
Vitamin A is needed for normal visual function and vitamin A deficiency (hypovitaminosis A) results in visual dysfunction that is reversible with vitamin A supplementation.
Several retinal diseases are directly or indirectly associated with vitamin A. Some are caused by pathogenic variants in genes encoding proteins that are directly involved in the vitamin A pathways in the retina, leading to local hypovitaminosis and/or accumulation of toxic bisretinoids. Other involve visual dysfunction due to malfunction of the visual pigment or local deficiency of vitamin A due to indirectly impaired transport of vitamin A from choroid to the retina (
Figure 1). Treatment with vitamin A in these cases is not straightforward, as there is a fear of accumulation of excessive amounts of toxic retinoids that are formed as byproducts of vitamin A pathways. A good example of this toxicity is ABCA4-retinopathy where toxic products of vitamin A accumulate excessively in the retina
[1][2][3][4][5].
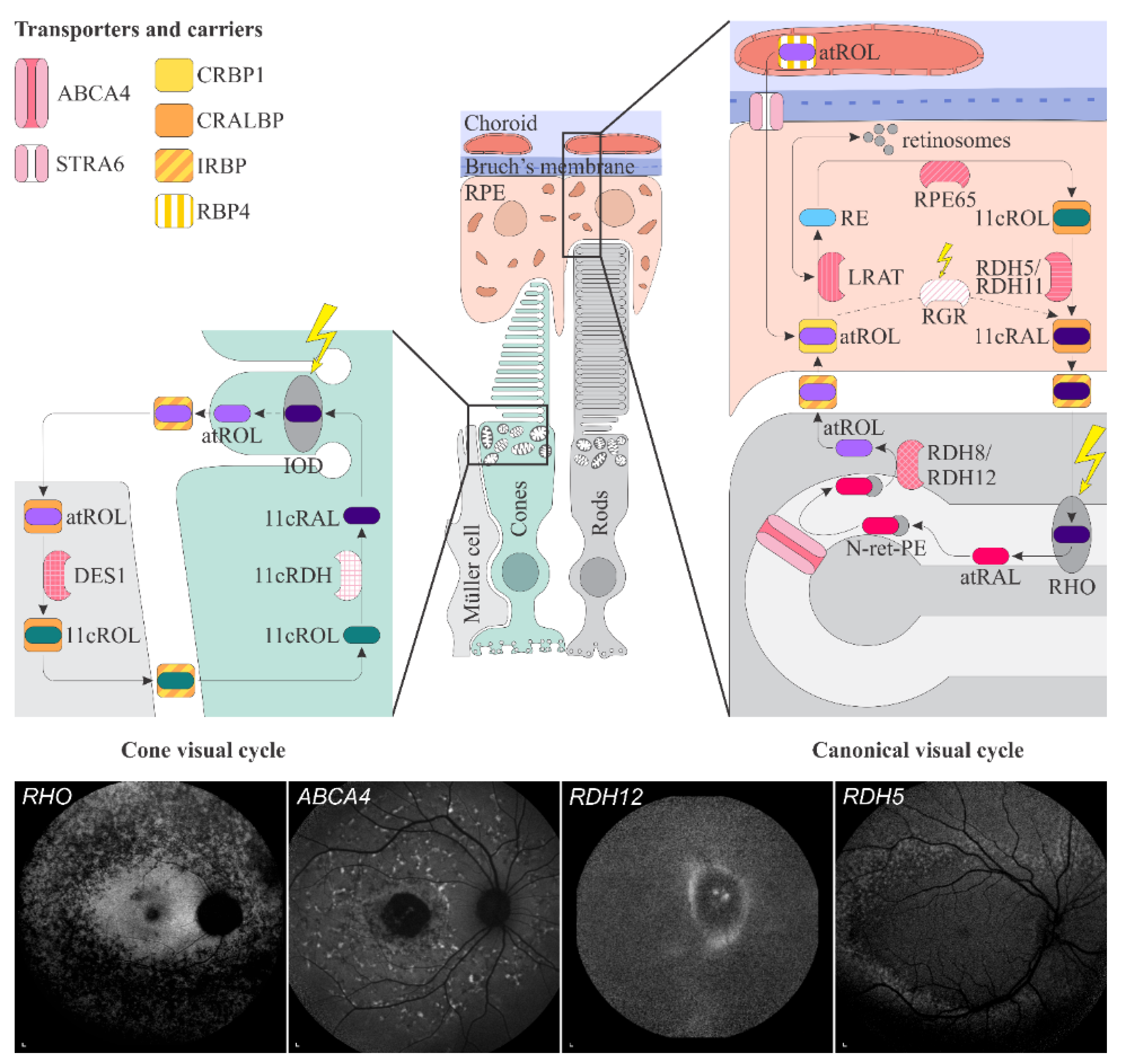 Figure 1.
Figure 1. Schematic representation of the canonical and cone visual cycles. Key enzymes, transporters, carriers and retinoids are shown. Fundus autofluorescence (FAF) images of representative patients, harbouring pathogenic variants in genes, encoding four essential proteins involved in the visual cycle, are presented below. Scale bars for four FAF images: 200 µm. Abbreviation explanation: RPE–retinal pigment epithelium, RBP4–retinol-binding protein 4, STRA6–stimulated by retinoic acid 6, CRBP1–cellular retinol-binding protein 1, LRAT–retinol:lecithin acyltransferase, RPE65–RPE-specific 65 kDa protein, RDH5–11-cis-retinol dehydrogenase 5, RDH11–11-cis-retinol dehydrogenase 11, CRALBP–cellular retinaldehyde-binding protein, RGR–retinal G protein-coupled receptor, IRBP–interphotoreceptor retinoid-binding protein, RDH8–all-trans-retinol dehydrogenase 8, RDH12–all-trans-retinol dehydrogenase 12, ABCA4–ATP-binding cassette subfamily A member 4, DES1–dihydroceramide desaturase-1, 11cRDH–11-cis-retinol dehydrogenase, RHO–rhodopsin, IOD–iodopsin, 11cRAL–11-cis-retinal, 11cROL–11-cis-retinol, atRAL–all-trans-retinal, atROL–all-trans-retinol, RE–retinyl esters, N-ret-PE–N-retinylidene-phosphatidylethanolamine, PE–phosphatidylethanolamine, A2E–N-retinyl-N-retinylidene ethanolamine. Source: Eye Hospital, University Medical Centre Ljubljana.
1.1. Retinal Signs of Hypovitaminosis A
Vitamin A deficiency in the retina primarily affects the rods, which results in night blindness, followed by cone dysfunction and impairment of daytime vision, including visual acuity. The delayed impairment of cones is thought to be due to their additional pathway for production of 11-cis-retinal in Müller cells
[6].
The functional impairment may be demonstrated using electrophysiology
[7][8][9]. Interestingly, S-cones were shown to be affected prior to the L and M cones
[8]. Recently, optical coherence tomography (OCT) has been shown as a useful diagnostic method, which shows abnormalities at the level of photoreceptor outer segments
[10] (
Figure 2). Similarly, vitamin A deficiency can be monitored by fundus autofluorescence (FAF) imaging. It may reveal hypoautofluorescent lesions
[11][12] and decreased autofluorescence signal
[13][14][15]. Reduced FAF can also be found in retinal diseases with impaired function of enzymes, involved in the conversion of all-trans-retinol into 11-cis-retinal in the retinal pigment epithelium (RPE) cells. These enzymes include RPE-specific 65 kDa protein (RPE65), retinol:lecithin acyltransferase (LRAT), cellular retinaldehyde-binding protein (CRALBP), 11-cis-retinol dehydrogenase 5 (RDH5) and 11-cis-retinol dehydrogenase 11 (RDH11)
[14][16]. Vitamin A replacement results in improvement of retinal function as well as structure, however long-term deficiency can lead to permanent degeneration of photoreceptors
[17][7][8][10][11][12][13][14][15][18]. Interestingly, hypovitaminosis A has also been associated with reversible ganglion cell thinning
[10], the reason for which is not understood but may reflect other vitamin A roles in the retina.
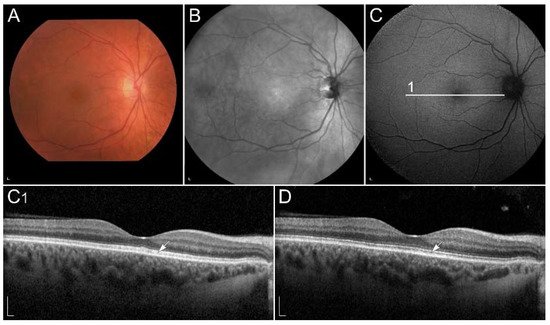
Figure 2. (A) Colour fundus image, (B) fundus infrared image and (C) FAF image in a patient with vitamin A deficiency. Note the corresponding (C1, arrow) spectral-domain optical coherence tomography (SD-OCT) image with abnormalities in photoreceptor outer segments, which (D, arrow) normalized after treatment with vitamin A supplementation. Patient’s best corrected Snellen decimal visual acuity before treatment was 0.7 on the right eye and 0.6 on the left eye. After treatment visual acuity improved to 1.0 on the right eye and 0.9 on the left eye. Scale bars: 200 µm. Source: Eye Hospital, University Medical Centre Ljubljana.
1.2. Retinal Diseases Associated with Pathogenic Variants in Genes Encoding Proteins Involved with Visual Cycle or Phototransduction
1.2.1. ABCA4-Retinopathy
Stargardt disease (STGD1), also known as fundus flavimaculatus or ABCA4-retinopathy, is a progressive disorder of the retina caused by bi-allelic variants in the gene that encodes ATP-binding cassette subfamily A member 4 (ABCA4), localized on chromosome 1p22.1. It is the most frequent retinal dystrophy caused by a single gene and is believed to affect approximately 1 in 8000–10,000 individuals worldwide
[19]. ABCA4 protein is a 250 kDa flippase, which transports all-trans-retinal across the photoreceptor membrane to the cytoplasmic side, making it an important part of the visual cycle and removal of toxic vitamin A products
[20][21].
ABCA4 dysfunction leads to the accumulation of vitamin A derivatives and toxic compounds, as they cannot be efficiently removed from the photoreceptor outer segment. Trapped vitamin A derivatives and toxic compounds can react nonenzymatically, leading to the formation of toxic bisretinoids. Upon phagocytosis of outer segments by adjacent RPE cells, the bisretinal compounds can be hydrolyzed by lysosomal enzymes to highly toxic metabolite A2E. The latter are insoluble, have a tendency to aggregate and accumulate as lipofuscin deposits in the RPE cells, compromising RPE function and leading to cell death. Consequently, dysfunction and loss of photoreceptors they support occur
[22][20][23].
In STGD1, cone degeneration is more severe and occurs before rod degeneration
[22][24]. The reason for that is not completely understood, however, it has been proposed that direct cone toxicity also plays a role in the disease pathogenesis, potentially due to the cone open lamellae
[24]. There are currently >1290 known pathogenic variants in the ABCA4 gene (
www.lovd.nl/ABCA4, accessed on 18 October 2021), causing a heterogeneous phenotypic appearance. Typically, the disease affects central vision, however, in more severe forms, it can also lead to blindness
[25]. Onset is most commonly in childhood or adolescence and least frequently in later adulthood, with a better prognosis usually associated with a later onset. Typical common clinical findings are central RPE atrophy, flecks and peripapillary sparing
[19][26] (
Figure 3).
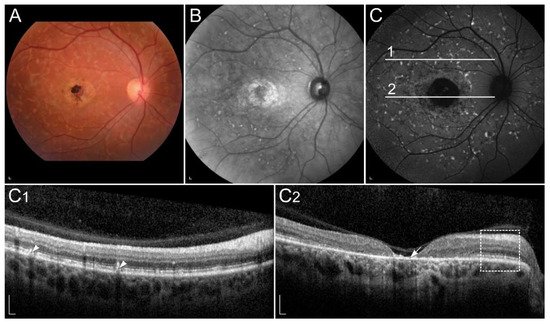
Figure 3. Clinical findings in a patient harbouring p.(Trp431*) and p.(Asp262Gly) in ABCA4. Macular affection, fundus flecks and peripapillary sparring (diagnostic triad) are shown on (A) colour fundus image, (B) fundus infrared image and (C) FAF image. On corresponding (C1,C2) SD-OCT images, (arrowheads) hyperautofluorescent flecks, (arrow) RPE atrophy and (rectangle) peripapillary sparing can also be observed. Patient’s best corrected Snellen decimal visual acuity was 0.1 on both eyes. Scale bars: 200 µm. Source: Eye Hospital, University Medical Centre Ljubljana.
Although ABCA4-retinopathy is caused by impaired function of a protein directly associated with vitamin A, there are only a few studies on the impact of vitamin A on its development
[27]. According to dietary questionnaire studies and animal model studies, high vitamin A intake is thought to be a risk factor for the progression
[3][28], therefore patients are discouraged from taking vitamin A supplements.
The vast majority of ongoing clinical trials (e.g., NCT03772665, NCT03364153, NCT02402660, NCT04239625) aim to target a particular step in the visual cycle and influence the process of A2E generation in various ways. However, these therapies are likely only to reduce symptoms of STGD1 and slower the rate of disease progression and not be curative
[29][30]. An example is slowing the visual cycle by inhibiting RPE65 or RBP4, which deprives photoreceptors of 11-cis-retinal and consequently reduces the accumulation of toxic A2E. As a side effect, reversible night blindness occurs
[31][32][33][34]. Alternative approach that does not compromise retinal function has been the provision of vitamin A deuterated at the carbon 20 position, which impedes the dimerization rate of vitamin A
[35][36][37]. However, no therapy has yet been registered.
1.2.2. Retinopathy Due to Pathogenic Variants in RPE65
RPE65 gene is located on chromosome 1p31.3 and encodes 65 kDa enzyme. RPE65 catalyses the conversion of all-trans-retinyl ester to 11-cis-retinol, allowing it to be reused in phototransduction and preventing its conversion to potentially toxic molecules. Pathogenic variants in RPE65 result in 11-cis-retinol and subsequently 11-cis-retinal deficiency, which essentially stops the visual cycle. In addition, retinyl esters accumulate in the RPE, leading to its damage
[38][39][40][41]. The pathogenesis, therefore, combines local vitamin A deficiency and its impaired metabolism, which leads to the formation of toxic byproducts.
Patients with pathogenic variants in the RPE65 gene have been presented with autosomal recessive RP, fundus albipunctatus, cone-rod dystrophy, and, most frequently, Leber congenital amaurosis (LCA) and early-onset severe retinal dystrophy (EOSRD) (LCA2, OMIM # 204100)
[42]. Pathogenic variants in RPE65 gene account for approximately 3% to 16% of LCA and EOSRD patients
[43]. LCA and EOSRD are characterized by severe visual loss from birth or early infancy, wandering nystagmus, amaurotic pupils, and markedly reduced or non-recordable full-field electroretinograms
[43][44]. Rod function is primarily affected, leading by cone dysfunction
[45]. The fundus appearance is initially normal, followed by the development of pigmentary changes over time. OCT reveals thinning of the outer nuclear layer, while FAF images show low signal due to reduced lipofuscin accumulation in the RPE. Lipofuscin cannot form as a result of a non-functional visual cycle
[46][47][48]. Most individuals are legally blind by the age of 40
[43].
Replacement therapy of missing 11-cis-retinol by synthetic 9-cis-retinyl acetate (QLT091001) has shown promising results in animal models, as it slowed the rate of retinal degeneration and improved the retinal function
[49][50]. An open-label, prospective, multi-center, phase 1b clinical trial of oral administration of QLT091001 in patients with defective RPE65 and LRAT genes also demonstrated improvement in vision. No serious adverse events occurred. However, some patients noted headaches, photophobia, nausea, reduction in serum high density lipoprotein concentrations, and an increase in serum triglycerides, alanine aminotransferase and aspartate aminotransferase concentrations due to high doses of vitamin A
[51][52].
These approaches have now been overshadowed by gene therapy for RPE65, that has been registered in 2017. The newly developed drug contains the active substance voretigene neparvovec. It employs adeno-associated virus vectors with genetically incorporated RPE65 cDNA. The introduction of a healthy RPE65 gene into the retina restores the normal visual cycle, which improves vision and prevents or at least delays retinal degeneration
[53][54][55].
1.2.3. Retinitis Pigmentosa Due to Pathogenic Variants in RHO
Retinitis pigmentosa (RP) is the most common progressive hereditary retinal degeneration, affecting approximately 1 in 4000 people
[56]. It is caused by pathogenic variants in almost 100 different genes and primarily affect rods. Since rods are responsible for low-light vision, night blindness is the first and characteristic manifestation of RP. In the later stage of the disease the cones are also affected, leading to visual field constriction and eventually central visual loss. Typical clinical findings, composing RP triad, are peripheral bone spicule pigmentation, attenuation of retinal vessels, and a waxy pallor of the optic nerve (
Figure 4A)
[56][57]. In most patients, FAF images show hyperautofluorescent ring, representing a transition zone between normal retinal function within the ring and absent outside of the ring. Hypoautofluorescent patches of RPE atrophy in the mid-periphery, related to a loss of peripheral vision, are normally seen in FAF images. Ring progressively constrict with time (
Figure 4C). The OCT image shows the perifoveal loss of the outer retina and the central preservation of the ellipsoid zone, which corresponds to the internal edges of the hyperautofluorescent ring visible on FAF (
Figure 4C1)
[56].
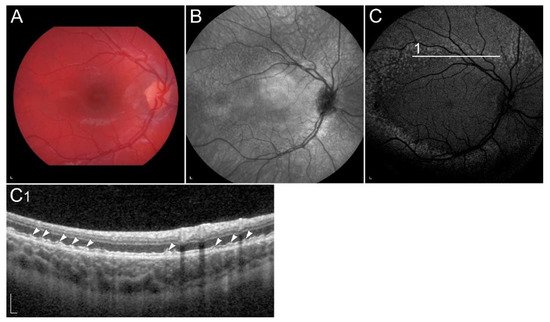
Figure 4. (A) Colour fundus image, (B) fundus infrared image and (C) FAF image with corresponding (C1) SD-OCT image showing clinical findings in a patient with p.(Gly90Asp) in RHO. Area within the arrowheads on C and C1 images corresponds to the preserved part of the retina. Patient’s best corrected Snellen decimal visual acuity was 0.6 on the right eye and 0.4 on the left eye. Scale bars: 200 µm. Source: Eye Hospital, University Medical Centre Ljubljana.
RP can be inherited in different patterns including X-linked, autosomal recessive, and autosomal dominant
[56][57]. The most common cause of autosomal dominant RP are pathogenic variants in the rhodopsin (RHO) gene that are responsible for 16% to 35% of cases with autosomal dominant RP
[58].
The vast majority of pathogenic variants in the RHO gene have been identified in patients with autosomal dominant RP, while rare pathogenic variants are also known to cause autosomal recessive RP or autosomal dominant congenital stationary night blindness
[58][59]. RHO was the first RP-linked gene identified.
[60]. It is located on chromosome 3q22.1, encoding 39 kDa highly photosensitive G protein-coupled receptor in rods
[57].
Clinical trial by Berson et al. reported that patients receiving 15,000 IU/d of vitamin A had a slower decline of 30-Hz ERG amplitude than those not receiving this dosage (p < 0.001), and therefore, retained more retinal function. Vitamin A was administered as retinyl palmitate. Patients were advised to maintain a regular diet without specifically selecting foods containing high levels of preformed vitamin A. The study included 601 patients (aged 18–49 years) with a treatment duration of 4 to 6 years
[61]. Besides vitamin A, it was also shown that long-chain omega-3 fatty acids
[62] and lutein intake slow the decline in visual function of patients with RP
[63][64]. However, vitamin A, long-chain omega-3 fatty acids and lutein supplementation have not yet been established in clinical practice for treating patients with RP, as long-term safety and efficacy of lutein and long-chain omega-3 fatty acids are still object of research, whereas vitamin A supplementation may cause side-effects.
1.2.4. Retinopathy Due to Pathogenic Variants in RDH5 or RDH11
RDH5 and RDH11 are thought to be the main 11-cis-retinol dehydrogenases in the RPE. While RDH5 is responsible for most of the activity in the RPE, RDH11 plays a minor role
[65].
RDH5 gene locates on chromosome 12q13-q14 and encodes 32 kDa membrane-associated protein
[66]. RDH5 catalyses the conversion of 11-cis-retinol to 11-cis-retinal through an NAD
+-dependent reaction in the RPE, which is the final oxidation step in the visual cycle
[67]. Pathogenic variants in RDH5 have been associated with autosomal recessive fundus albipunctatus, which belongs to a group of hereditary flecked retina syndromes
[68].
Fundus albipunctatus is a rare form of congenital stationary night blindness with delayed dark adaptation after exposure to bright light due to slowed production of 11-cis-retinal
[69][70]. Impaired 11-cis-retinal production also leads to decreased A2E and lipofuscin formation, which results in reduced autofluorescence in FAF images
[14][16][70][71][72]. It is characterised with stationary or slow progression of rod abnormalities, and numerous retinal flecks placed throughout the retina, except the fovea
[70][73]. Flecks are thought to represent an accumulation of toxic retinyl esters in the RPE (
Figure 5)
[74]. Around 30% of patients with fundus albipunctatus, usually elderly, develop also progressive cone dystrophy
[70][75].
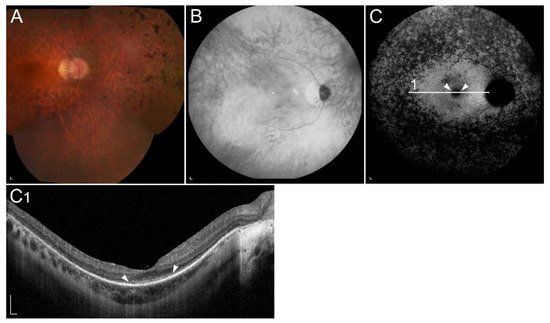
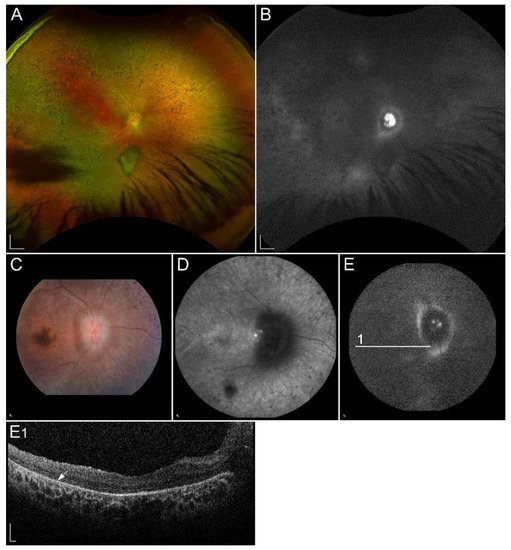
Figure 5. Clinical characteristics of a patient homozygous for p.(Thr137Ser) in RDH5. (A) Colour fundus image, (B) infrared image and (C) FAF image with corresponding (C1) SD-OCT image showing (arrowheads) retinal flecks. FAF image shows reduced autofluorescence in the entire retina. Patient’s best corrected Snellen decimal visual acuity was 0.8 on both eyes. Scale bars: 200 µm. Source: Eye Hospital, University Medical Centre Ljubljana.
Administration of 9-cis-β-carotene in a small group of patients with fundus albipunctatus showed promising results. However, no further studies on bigger cohorts were performed, and, therefore, 9-cis-β-carotene is not accepted as a treatment for patients with fundus albipunctatus
[76].
Recovery of night vision after prolonged dark adaptation suggests that RDH11 plays a complementary role to RDH5
[74]. Besides participating in the oxidation of 11-cis-retinol to 11-cis-retinal, RDH11 also catalyzes a reduction of 11-cis-retinal. Moreover, it has the ability to catalyze the oxidation of trans-retinol and reduction of trans-retinal
[77][65][78][79]. RDH11 has an approximate molecular mass of 35 kDa and is encoded by the RDH11 gene located on chromosome 14q24.1. Pathogenic variants in the RDH11 gene are associated with syndromic RP, which features include atypical RP, facial dysmorphologies, psychomotor developmental delay, learning disabilities and short stature
[80]. Decreased FAF has also been described, as lipofuscin formation is reduced
[14].
1.2.5. Retinopathy Due to Pathogenic Variants in RDH8 or RDH12
All-trans-retinol dehydrogenase 8 (RHD8) is located on chromosome 19p13.2, while all-trans-retinol dehydrogenase 12 (RDH12) locates on chromosome 14q24.1. RDH12, with a molecular mass of 35 kDa, and RDH8, with a molecular mass of about 34 kDa, are the major all-trans-retinal dehydrogenases in the photoreceptors and use NADPH as a cofactor
[81][77]. RDH8 is located in the outer segment of cones and rods, while RDH12 is in the inner segments of cones and rods. RDH8 contributes to approximately 70% and RDH12 to approximately 30% of all-trans-RDH activity. Their dysfunction subsequently leads to decreased synthesis of 11-cis-retinal, which may result in reduced autofluorescence signal
[81][82][77][83][84].
Pathogenic variants in RDH12 have been shown to cause autosomal recessive LCA and EOSRD (LCA13, OMIM #612712), characterized by severe progressive rod-cone dystrophy with widespread RPE atrophy, bone-spicule pigmentation, vascular attenuation, macular atrophy and corresponding macular excavation
[44][83][85][86]. A phenotypic feature of RDH12 retinopathy is sparing of the peripapillary region
[83][85][87] (
Figure 6), pathognomonic of ABCA4-retinopathy. Mutations in RDH12 gene have been found in patients with autosomal recessive retinitis RP, cone-rod dystrophy as well as macular dystrophy
[88]. Interestingly, pathogenic variants in RDH8 have not yet been linked to any retinal disease.
Figure 6. (A,C) Ultra-widefield and 50° colour fundus images, (D) fundus infrared image, (B,E) ultra-widefield and 55° FAF images and (E1) SD-OCT image of a patient homozygous for p.(Ala126Glu) in RDH12. (A,C) Colour fundus images and (D) fundus infrared image demonstrate bone-spicule pigmentation and wide-spread retinal atrophy. The latest is also shown on (E1) OCT, which shows loss of the photoreceptor inner segment ellipsoid band and (arrow) atrophy of the RPE. Peripapillary retinal preservation can be seen on A, B, C, D and E images. The quality of E and E1 images is low due to reduced visual acuity and nystagmus. Patient’s best corrected Snellen decimal visual acuity was 0.3 on both eyes. Scale bars A, B: 2 mm; C, D, E and E1: 200 µm. Source: Eye Hospital, University Medical Centre Ljubljana.
1.2.6. Retinopathy Due to Pathogenic Variants in RLBP1
The retinaldehyde-binding protein 1 (RLBP1) gene is localized on chromosome 15q26 and transcribes RLBP1 also known as CRALBP, abundant in the RPE and Müller cells
[82][89][90]. CRALBP is a 36 kDa water-soluble protein, that transports hydrophobic 11-cis-retinoids
[91][92][93][94]. Pathogenic variants in the RLBP1 gene prevent regeneration of 11-cis-retinal, which leads to reduced autofluorescence signal
[14][95]. They have been associated with a number of autosomal recessive retinal dystrophies, including RP, fundus albipunctatus, Bothnia dystrophy, Newfoundland rod-cone dystrophy and retinitis punctata albescens
[84][95][96]. The latter has been linked by pathogenic variants in five different genes, whereas the RLBP1 gene is the most frequently associated with retinitis punctata albescens
[97]. Retinitis punctata albescens is a rod-cone dystrophy subtype of RP and accounts for 0.5% of all RP cases, with a prevalence of 1 in 800,000
[98]. It is characterised by nyctalopia in early childhood and numerous punctate whitish-yellow spots throughout the entire fundus. Atrophy of the RPE appears mid-peripherally and progressively enlarge towards the centre. Macular involvement is frequent
[90][96][98][99]. Dark adaptation is always abnormal and occurs even prior to retinal degeneration
[95][98]. RLBP1 gene therapy clinical trial is currently ongoing (NCT03374657).
1.2.7. Retinopathy Due to Pathogenic Variants in RBP3
Interphotoreceptor retinoid-binding protein (IRBP), also known as retinol-binding protein 3 (RBP3), is a136 kDa lipoglycoprotein product of the RBP3 gene on chromosome 10q11.22
[100][101]. It is secreted by photoreceptors and accumulates in the interphotoreceptor matrix. It functions as the transporter of retinoids between the photoreceptors and RPE and also between photoreceptors and Müller cells
[102][103][104][105]. Pathogenic variants in RBP3 gene are described as an infrequent cause of autosomal recessive RP
[106]. They can also cause unusual retinal dystrophy characterized by childhood onset high myopia, generalized rod and cone dysfunction, and an insignificant fundus appearance
[107].
1.2.8. Retinopathy Due to Pathogenic Variants in RBP4
Retinol-binding protein 4 (RBP4) is a specific carrier for all-trans-retinol in the circulation, encoded by the RBP4 gene, which is located on chromosome 10q23.33
[101]. To prevent extensive kidney filtration and lowered plasma concentrations of RBP4, holo-RBP4 associates with TTR. Therefore, the formation of holo-RBP:TTR complex represents an important step in vitamin A homeostasis
[108][109]. Pathogenic variants in RBP4 gene lead to reduced plasma RBP4 and all-trans-retinol concentrations and are associated with night blindness
[110][111][112][113], microphthalmia
[113][114], anophthalmia
[114], coloboma
[110][112][114], retinal dystrophy
[110][111][112][113][115] and acne vulgaris
[110][112][115].
1.2.9. Retinopathy Due to Pathogenic Variants in RGR
Retinal G protein-coupled receptor (RGR) gene is located on chromosome 10q23.1
[116] and encodes a 28 kDa intracellular opsin localized to RPE and Müller cells
[117][118]. As the association of variants in RGR gene with specific ocular diseases has been rarely reported
[119][120], clinical data are very limited. Spectrum of retinal changes in published cases ranged from normal visual acuity, normal electroretinogram and diffuse or reticular pigmentation of the retina to severe visual loss with diffuse atrophy of the retina and choroid
[121].
1.2.10. Retinopathy Due to Pathogenic Variants in LRAT
LRAT is a 36 kDa protein encoded by LRAT gene located on chromosome 4q32.1
[122]. It converts all-trans-retinol to all-trans-retinyl esters. Pathogenic variants in LRAT gene cause LCA and RP
[123][124][125]. Similarly to pathogenic variants in the RPE65 gene, pathogenic variants in LRAT also lead to 11-cis-retinal deficiency and, therefore, lack of autofluorescence
[14][125]. Treatment with oral synthetic cis-retinoid QLT091001 showed promising results in patients. However, the adverse effects always have to be a concern
[51][52].
1.2.11. Retinopathy Due to Pathogenic Variants in STRA6
Stimulated by retinoic acid 6 (STRA6) gene is located on chromosome 15q24.1 and encodes transmembrane receptor
[126], with a molecular mass of 74 kDa
[127]. It catalyses the release of retinol from RBP4 and facilitates its translocation across the RPE cell membrane to the cytosol
[128][126]. STRA6 gene pathogenic variants are associated with Matthew-Wood syndrome, which is in eyes presented with anophthalmia
[82][126][129][130][131][132].
1.3. Retinal Diseases Involving Local Vitamin A Deficiency
1.3.1. Sorsby Fundus Dystrophy
Sorsby fundus dystrophy (SFD) is rare retinal dystrophy with autosomal dominant pattern of inheritance. The estimated prevalence is 1 in 220,000
[133]. It is caused by pathogenic variants in the gene encoding a tissue inhibitor of metalloproteinases-3 (TIMP3), located on chromosome 22q12.3
[134] which encodes a 24 kDa glycoprotein
[133]. TIMP3 is mainly expressed and secreted by the RPE and is an element of Bruch’s membrane. It regulates turnover of the extracellular matrix, inflammation and possesses pro-apoptotic and anti-angiogenic activities
[4][135].
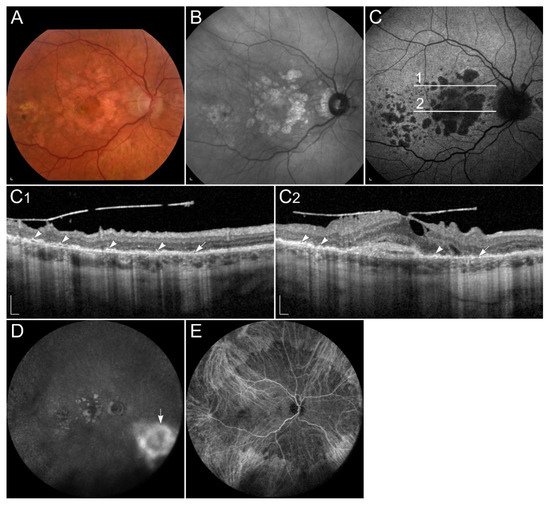
Pathogenic variants in the TIMP3 gene result in thickening of the Bruch’s membrane, leading to reduced exchange of nutrients and waste products between RPE and choriocapillaris, including vitamin A, resulting in local vitamin A deficiency
[4][133]. Symptoms usually appear after the 2nd decade of life, with an average onset in the 4th to 6th decade
[136]. Typically, the earliest symptom is night blindness. However, later the disease is often complicated with choroidal neovascularizations, leading to metamorphopsia, reduced colour vision, and loss of central vision. Clinically, the disease manifests with thickened Bruch’s membrane, drusen-like deposits in the sub-RPE space, and choroidal neovascularization, which usually requires treatment with vascular endothelial growth factor (VEGF) inhibitors. Later, expanding areas of geographic atrophy may also develop
[4][133][135][137][138][139] (
Figure 7). Indocyanine green angiography can show blockade of choroidal fluorescence by the thickened Bruch membrane
[135][140] (
Figure 7E).
Figure 7. Patient with Sorsby dystrophy due to pathogenic variant p.(Ser170Cys) in TIMP3. (A) Colour fundus image, (B) fundus infrared image, and (C) FAF image with corresponding (C1,C2) SD-OCT images showing (arrows) geographic atrophy and (arrowheads) subretinal drusen-like deposits. (D, arrow) Hyperfluorescent area is compatible with choroidal neovascularisation. (E) The reduced indocyanine green late-phase angiography fluorescence is due to decreased permeability of Bruch’s membrane. Patient’s best corrected Snellen decimal visual acuity was 0.4 on the right eye and 0.6 on the left eye. Scale bars: 200 µm. Source: Eye Hospital, University Medical Centre Ljubljana.
Early attempts of SFD treatment included adding vitamin A to improve symptoms of night blindness. Although initial results were promising, treatment did not become widely used due to the potential long-term toxicity of high doses of vitamin A and lack of lower doses efficacy
[4][135].
2. Summary of the Relevant Clinical Trials
There are many different preclinical and clinical trials where researchers try to elucidate the mechanisms and develop effective treatments for retinal diseases associated with vitamin A. Some of them include targeted gene therapy, while others include non-genetic therapies affecting the visual cycle. The most relevant trials for each disease are stated in each chapter above and summarized in Table 1.
Table 1. Summary of the relevant clinical trials.
| NCT Number |
Disease |
Drug |
Sponsor |
Number of Subjects |
Phase of the Study |
Mechanism |
| NCT03772665 |
STGD1 |
Emixustat (inhibitor of RPE65) |
Kubota Vision Inc. |
194 |
3 |
Slower regeneration of 11-cis-retinal. |
| NCT03364153 |
STGD1 |
Zimura (anti-C5 aptamer) |
IVERIC bio, Inc. |
120 |
2 |
Prevention of the destructive effects of the activated complement cascade. |
| NCT02402660 and NCT04239625 |
STGD1 |
ALK-001 (C20-deuterated vitamin A) |
Alkeus Pharmaceuticals, Inc. |
140 |
2 |
Impaired dimerization of vitamin A and therefore reduced production of A2E. |
| NCT03374657 |
RP |
CPK850 (RLBP1 promoter) |
Novartis Pharmaceuticals |
Recruiting |
1 and 2 |
Gene therapy. |
| NCT03478865 and NCT03478878 |
AMD |
Vitamin A palmitate |
National Eye Institute (NEI) |
Recruiting |
1 |
Vitamin A supplementation. |
Abbreviation explanation: AMD–age-related macular degeneration. Source:
ClinicalTrials.gov, accessed on 7 January 2022.
 +1 credit
+1 credit