Vitamin A is an essential fat-soluble vitamin that occurs in various chemical forms. It is essential for several physiological processes. Either hyper- or hypovitaminosis can be harmful. One of the most important vitamin A functions is its involvement in visual phototransduction, where it serves as the crucial part of photopigment, the first molecule in the process of transforming photons of light into electrical signals. In this process, large quantities of vitamin A in the form of 11-cis-retinal are being isomerized to all-trans-retinal and then quickly recycled back to 11-cis-retinal. Complex machinery of transporters and enzymes is involved in this process (i.e., the visual cycle). Any fault in the machinery may not only reduce the efficiency of visual detection but also cause the accumulation of toxic chemicals in the retina.
- vitamin A
- visual cycle
- retinal diseases
- ABCA4
- RHO
- RDH5
- RDH12
- treatment
1. Retinal Diseases Directly or Indirectly Associated with Vitamin A and Its Pathways
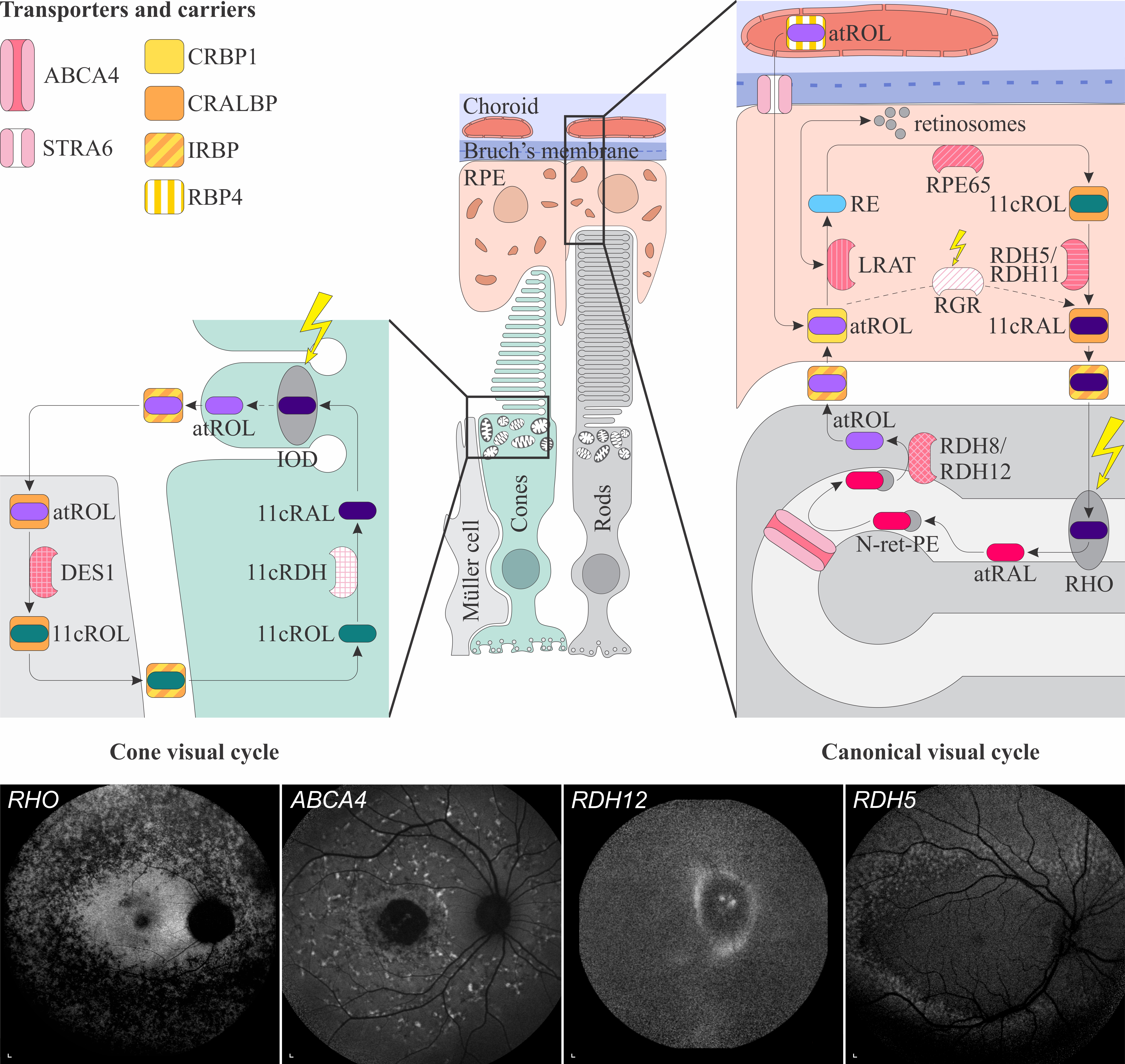
Figure 1. Schematic representation of the canonical and cone visual cycles. Key enzymes, transporters, carriers and retinoids are shown. Fundus autofluorescence (FAF) images of representative patients, harbouring pathogenic variants in genes, encoding four essential proteins involved in the visual cycle, are presented below. Scale bars for four FAF images: 200 µm. Abbreviation explanation: RPE–retinal pigment epithelium, RBP4–retinol-binding protein 4, STRA6–stimulated by retinoic acid 6, CRBP1–cellular retinol-binding protein 1, LRAT–retinol:lecithin acyltransferase, RPE65–RPE-specific 65 kDa protein, RDH5–11-cis-retinol dehydrogenase 5, RDH11–11-cis-retinol dehydrogenase 11, CRALBP–cellular retinaldehyde-binding protein, RGR–retinal G protein-coupled receptor, IRBP–interphotoreceptor retinoid-binding protein, RDH8–all-trans-retinol dehydrogenase 8, RDH12–all-trans-retinol dehydrogenase 12, ABCA4–ATP-binding cassette subfamily A member 4, DES1–dihydroceramide desaturase-1, 11cRDH–11-cis-retinol dehydrogenase, RHO–rhodopsin, IOD–iodopsin, 11cRAL–11-cis-retinal, 11cROL–11-cis-retinol, atRAL–all-trans-retinal, atROL–all-trans-retinol, RE–retinyl esters, N-ret-PE–N-retinylidene-phosphatidylethanolamine, PE–phosphatidylethanolamine, A2E–N-retinyl-N-retinylidene ethanolamine. Source: Eye Hospital, University Medical Centre Ljubljana.
1.1. Retinal Signs of Hypovitaminosis A
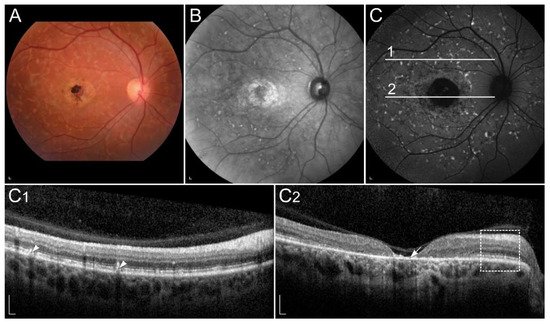
1.2. Retinal Diseases Associated with Pathogenic Variants in Genes Encoding Proteins Involved with Visual Cycle or Phototransduction
1.2.1. ABCA4-Retinopathy
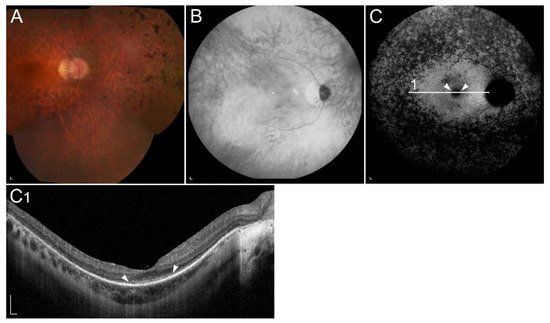
1.2.2. Retinopathy Due to Pathogenic Variants in RPE65
1.2.3. Retinitis Pigmentosa Due to Pathogenic Variants in RHO
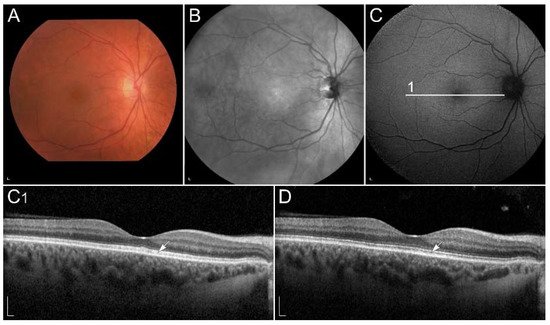
1.2.4. Retinopathy Due to Pathogenic Variants in RDH5 or RDH11
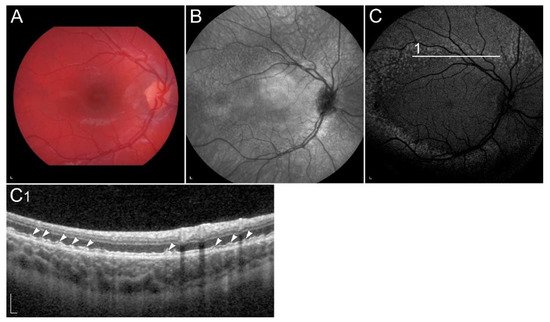
1.2.5. Retinopathy Due to Pathogenic Variants in RDH8 or RDH12
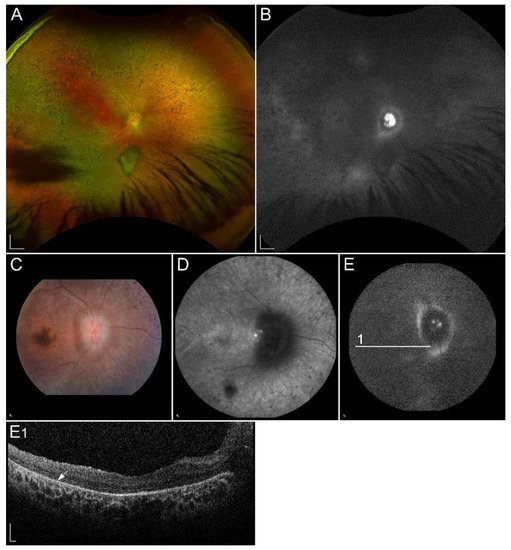
1.2.6. Retinopathy Due to Pathogenic Variants in RLBP1
1.2.7. Retinopathy Due to Pathogenic Variants in RBP3
1.2.8. Retinopathy Due to Pathogenic Variants in RBP4
1.2.9. Retinopathy Due to Pathogenic Variants in RGR
1.2.10. Retinopathy Due to Pathogenic Variants in LRAT
1.2.11. Retinopathy Due to Pathogenic Variants in STRA6
1.3. Retinal Diseases Involving Local Vitamin A Deficiency
1.3.1. Sorsby Fundus Dystrophy
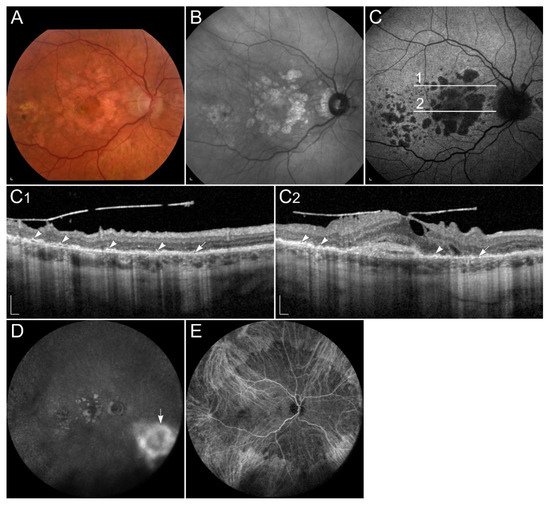
References
- Álvarez, R.; Vaz, B.; Gronemeyer, H.; de Lera, Á.R. Functions, therapeutic applications, and synthesis of retinoids and carotenoids. Chem. Rev. 2014, 114, 1–125.
- Chew, E.Y.; Clemons, T.E.; Sangiovanni, J.P.; Danis, R.P.; Ferris, F.L., 3rd; Elman, M.J.; Antoszyk, A.N.; Ruby, A.J.; Orth, D.; Bressler, S.B.; et al. Secondary analyses of the effects of lutein/zeaxanthin on age-related macular degeneration progression: AREDS2 report No. 3. JAMA Ophthalmol. 2014, 132, 142–149.
- Sofi, F.; Sodi, A.; Franco, F.; Murro, V.; Biagini, D.; Miele, A.; Abbruzzese, G.; Mucciolo, D.P.; Virgili, G.; Menchini, U.; et al. Dietary profile of patients with Stargardt’s disease and Retinitis Pigmentosa: Is there a role for a nutritional approach? BMC Ophthalmol. 2016, 16, 13.
- Anand-Apte, B.; Chao, J.R.; Singh, R.; Stöhr, H. Sorsby fundus dystrophy: Insights from the past and looking to the future. J. Neurosci. Res. 2019, 97, 88–97.
- Brito-García, N.; Del Pino-Sedeño, T.; Trujillo-Martín, M.M.; Coco, R.M.; Rodríguez de la Rúa, E.; Del Cura-González, I.; Serrano-Aguilar, P. Effectiveness and safety of nutritional supplements in the treatment of hereditary retinal dystrophies: A systematic review. Eye 2017, 31, 273–285.
- Miyazono, S.; Shimauchi-Matsukawa, Y.; Tachibanaki, S.; Kawamura, S. Highly efficient retinal metabolism in cones. Proc. Natl. Acad. Sci. USA 2008, 105, 16051–16056.
- Saker, S.; Morales, M.; Jhittay, H.; Wen, Y.; Amoaku, W. Electrophysiological and microperimetry changes in vitamin A deficiency retinopathy. Doc. Ophthalmol. 2015, 130, 231–240.
- McBain, V.A.; Egan, C.A.; Pieris, S.J.; Supramaniam, G.; Webster, A.R.; Bird, A.C.; Holder, G.E. Functional observations in vitamin A deficiency: Diagnosis and time course of recovery. Eye 2007, 21, 367–376.
- Kakiuchi, D.; Uehara, T.; Shiotani, M.; Nakano-Ito, K.; Suganuma, A.; Aoki, T.; Tsukidate, K.; Sawada, K. Oscillatory potentials in electroretinogram as an early marker of visual abnormalities in vitamin A deficiency. Mol. Med. Rep. 2015, 11, 995–1003.
- Jevnikar, K.; Šuštar, M.; Kozjek, N.R.; Štrucl, A.M.; Markelj, Š.; Hawlina, M.; Fakin, A. Disruption of the outer segments of the photoreceptors on OCT as a feature of vitamin A deficiency. Retin. Cases Brief Rep. 2020.
- Aleman, T.S.; Garrity, S.T.; Brucker, A.J. Retinal structure in vitamin A deficiency as explored with multimodal imaging. Doc. Ophthalmol. 2013, 127, 239–243.
- Apushkin, M.A.; Fishman, G.A. Improvement in visual function and fundus findings for a patient with vitamin A-deficient retinopathy. Retina 2005, 25, 650–652.
- Lima de Carvalho, J.R., Jr.; Tsang, S.H.; Sparrow, J.R. Vitamin a Deficiency Monitored by Quantitative Short Wavelength Fundus Autofluorescence in a Case of Bariatric Surgery. Retin. Cases Brief Rep. 2019.
- Oh, J.K.; Lima de Carvalho, J.R.; Ryu, J.; Tsang, S.H.; Sparrow, J.R. Short-Wavelength and Near-Infrared Autofluorescence in Patients with Deficiencies of the Visual Cycle and Phototransduction. Sci. Rep. 2020, 10, 8998.
- Braunstein, A.; Trief, D.; Wang, N.-K.; Chang, S.; Tsang, S.H. Vitamin A deficiency in New York City. Lancet 2010, 376, 267.
- Wang, N.K.; Chuang, L.H.; Lai, C.C.; Chou, C.L.; Chu, H.Y.; Yeung, L.; Chen, Y.P.; Chen, K.J.; Wu, W.C.; Chen, T.L.; et al. Multimodal fundus imaging in fundus albipunctatus with RDH5 mutation: A newly identified compound heterozygous mutation and review of the literature. Doc. Ophthalmol. 2012, 125, 51–62.
- Zhong, M.; Kawaguchi, R.; Kassai, M.; Sun, H. Retina, retinol, retinal and the natural history of vitamin A as a light sensor. Nutrients 2012, 4, 2069–2096.
- Owsley, C.; McGwin, G.; Jackson, G.R.; Heimburger, D.C.; Piyathilake, C.J.; Klein, R.; White, M.F.; Kallies, K. Effect of short-term, high-dose retinol on dark adaptation in aging and early age-related maculopathy. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1310–1318.
- Cremers, F.P.M.; Lee, W.; Collin, R.W.J.; Allikmets, R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog. Retin. Eye Res. 2020, 79, 100861.
- Molday, R.S. Insights into the Molecular Properties of ABCA4 and Its Role in the Visual Cycle and Stargardt Disease. Prog. Mol. Biol. Transl. Sci. 2015, 134, 415–431.
- Tsybovsky, Y.; Molday, R.S.; Palczewski, K. The ATP-binding cassette transporter ABCA4: Structural and functional properties and role in retinal disease. Adv. Exp. Med. Biol. 2010, 703, 105–125.
- Molday, R.S.; Zhong, M.; Quazi, F. The role of the photoreceptor ABC transporter ABCA4 in lipid transport and Stargardt macular degeneration. Biochim. Biophys. Acta 2009, 1791, 573–583.
- Quazi, F.; Molday, R.S. ATP-binding cassette transporter ABCA4 and chemical isomerization protect photoreceptor cells from the toxic accumulation of excess 11-cis-retinal. Proc. Natl. Acad. Sci. USA 2014, 111, 5024–5029.
- Conley, S.M.; Cai, X.; Makkia, R.; Wu, Y.; Sparrow, J.R.; Naash, M.I. Increased cone sensitivity to ABCA4 deficiency provides insight into macular vision loss in Stargardt’s dystrophy. Biochim. Biophys. Acta 2012, 1822, 1169–1179.
- Fakin, A.; Robson, A.G.; Fujinami, K.; Moore, A.T.; Michaelides, M.; Holder, G.E.; Webster, A.R. Phenotype and Progression of Retinal Degeneration Associated with Nullizigosity of ABCA4. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4668–4678.
- Tanna, P.; Strauss, R.W.; Fujinami, K.; Michaelides, M. Stargardt disease: Clinical features, molecular genetics, animal models and therapeutic options. Br. J. Ophthalmol. 2017, 101, 25–30.
- Federspiel, C.A.; Bertelsen, M.; Kessel, L. Vitamin A in Stargardt disease-an evidence-based update. Ophthalmic Genet. 2018, 39, 555–559.
- Radu, R.A.; Yuan, Q.; Hu, J.; Peng, J.H.; Lloyd, M.; Nusinowitz, S.; Bok, D.; Travis, G.H. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following Vitamin A supplementation. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3821–3829.
- Piotter, E.; McClements, M.E.; MacLaren, R.E. Therapy Approaches for Stargardt Disease. Biomolecules 2021, 11, 1179.
- Hussain, R.M.; Ciulla, T.A.; Berrocal, A.M.; Gregori, N.Z.; Flynn, H.W.; Lam, B.L. Stargardt macular dystrophy and evolving therapies. Expert Opin. Biol. Ther. 2018, 18, 1049–1059.
- Marmor, M.F.; Jain, A.; Moshfeghi, D. Total rod ERG suppression with high dose compassionate Fenretinide usage. Doc. Ophthalmol. 2008, 117, 257–261.
- Zhang, J.; Kiser, P.D.; Badiee, M.; Palczewska, G.; Dong, Z.; Golczak, M.; Tochtrop, G.P.; Palczewski, K. Molecular pharmacodynamics of emixustat in protection against retinal degeneration. J. Clin. Investig. 2015, 125, 2781–2794.
- Dobri, N.; Qin, Q.; Kong, J.; Yamamoto, K.; Liu, Z.; Moiseyev, G.; Ma, J.X.; Allikmets, R.; Sparrow, J.R.; Petrukhin, K. A1120, a nonretinoid RBP4 antagonist, inhibits formation of cytotoxic bisretinoids in the animal model of enhanced retinal lipofuscinogenesis. Investig. Ophthalmol. Vis. Sci. 2013, 54, 85–95.
- Mata, N.L.; Lichter, J.B.; Vogel, R.; Han, Y.; Bui, T.V.; Singerman, L.J. Investigation of oral fenretinide for treatment of geographic atrophy in age-related macular degeneration. Retina 2013, 33, 498–507.
- Charbel Issa, P.; Barnard, A.R.; Herrmann, P.; Washington, I.; MacLaren, R.E. Rescue of the Stargardt phenotype in Abca4 knockout mice through inhibition of vitamin A dimerization. Proc. Natl. Acad. Sci. USA 2015, 112, 8415–8420.
- Kaufman, Y.; Ma, L.; Washington, I. Deuterium enrichment of vitamin A at the C20 position slows the formation of detrimental vitamin A dimers in wild-type rodents. J. Biol. Chem. 2011, 286, 7958–7965.
- Ma, L.; Kaufman, Y.; Zhang, J.; Washington, I. C20-D3-vitamin A slows lipofuscin accumulation and electrophysiological retinal degeneration in a mouse model of Stargardt disease. J. Biol. Chem. 2011, 286, 7966–7974.
- Redmond, T.M. RPE65 takes on another role in the vertebrate retina. Proc. Natl. Acad. Sci. USA 2017, 114, 10818–10820.
- Cai, X.; Conley, S.M.; Naash, M.I. RPE65: Role in the visual cycle, human retinal disease, and gene therapy. Ophthalmic Genet. 2009, 30, 57–62.
- Cideciyan, A.V. Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy. Prog. Retin. Eye Res. 2010, 29, 398–427.
- Takahashi, Y.; Moiseyev, G.; Nikolaeva, O.; Ma, J.-x. Identification of the key residues determining the product specificity of isomerohydrolase. Biochemistry 2012, 51, 4217–4225.
- Li, S.; Xiao, X.; Yi, Z.; Sun, W.; Wang, P.; Zhang, Q. RPE65 mutation frequency and phenotypic variation according to exome sequencing in a tertiary centre for genetic eye diseases in China. Acta Ophthalmol. 2020, 98, e181–e190.
- Aoun, M.; Passerini, I.; Chiurazzi, P.; Karali, M.; De Rienzo, I.; Sartor, G.; Murro, V.; Filimonova, N.; Seri, M.; Banfi, S. Inherited Retinal Diseases Due to RPE65 Variants: From Genetic Diagnostic Management to Therapy. Int. J. Mol. Sci. 2021, 22, 7207.
- Kumaran, N.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 2017, 101, 1147–1154.
- Jacobson, S.G.; Aleman, T.S.; Cideciyan, A.V.; Roman, A.J.; Sumaroka, A.; Windsor, E.A.; Schwartz, S.B.; Heon, E.; Stone, E.M. Defining the residual vision in leber congenital amaurosis caused by RPE65 mutations. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2368–2375.
- Lorenz, B.; Wabbels, B.; Wegscheider, E.; Hamel, C.P.; Drexler, W.; Preising, M.N. Lack of fundus autofluorescence to 488 nanometers from childhood on in patients with early-onset severe retinal dystrophy associated with mutations in RPE65. Ophthalmology 2004, 111, 1585–1594.
- Kumaran, N.; Georgiou, M.; Bainbridge, J.W.B.; Bertelsen, M.; Larsen, M.; Blanco-Kelly, F.; Ayuso, C.; Tran, H.V.; Munier, F.L.; Kalitzeos, A.; et al. Retinal Structure in RPE65-Associated Retinal Dystrophy. Investig. Ophthalmol. Vis. Sci. 2020, 61, 47.
- Katz, M.L.; Redmond, T.M. Effect of Rpe65 knockout on accumulation of lipofuscin fluorophores in the retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2001, 42, 3023–3030.
- Van Hooser, J.P.; Aleman, T.S.; He, Y.-G.; Cideciyan, A.V.; Kuksa, V.; Pittler, S.J.; Stone, E.M.; Jacobson, S.G.; Palczewski, K. Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proc. Natl. Acad. Sci. USA 2000, 97, 8623.
- Maeda, T.; Dong, Z.; Jin, H.; Sawada, O.; Gao, S.; Utkhede, D.; Monk, W.; Palczewska, G.; Palczewski, K. QLT091001, a 9-cis-retinal analog, is well-tolerated by retinas of mice with impaired visual cycles. Investig. Ophthalmol. Vis. Sci. 2013, 54, 455–466.
- Scholl, H.P.N.; Moore, A.T.; Koenekoop, R.K.; Wen, Y.; Fishman, G.A.; van den Born, L.I.; Bittner, A.; Bowles, K.; Fletcher, E.C.; Collison, F.T.; et al. Safety and Proof-of-Concept Study of Oral QLT091001 in Retinitis Pigmentosa Due to Inherited Deficiencies of Retinal Pigment Epithelial 65 Protein (RPE65) or Lecithin:Retinol Acyltransferase (LRAT). PLoS ONE 2015, 10, e0143846.
- Koenekoop, R.K.; Sui, R.; Sallum, J.; van den Born, L.I.; Ajlan, R.; Khan, A.; den Hollander, A.I.; Cremers, F.P.; Mendola, J.D.; Bittner, A.K.; et al. Oral 9-cis retinoid for childhood blindness due to Leber congenital amaurosis caused by RPE65 or LRAT mutations: An open-label phase 1b trial. Lancet 2014, 384, 1513–1520.
- Wang, X.; Yu, C.; Tzekov, R.T.; Zhu, Y.; Li, W. The effect of human gene therapy for RPE65-associated Leber’s congenital amaurosis on visual function: A systematic review and meta-analysis. Orphanet J. Rare Dis. 2020, 15, 49.
- Miraldi Utz, V.; Coussa, R.G.; Antaki, F.; Traboulsi, E.I. Gene therapy for RPE65-related retinal disease. Ophthalmic Genet. 2018, 39, 671–677.
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860.
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186.
- Daiger, S.P.; Sullivan, L.S.; Bowne, S.J. Genes and mutations causing retinitis pigmentosa. Clin. Genet. 2013, 84, 132–141.
- Beryozkin, A.; Levy, G.; Blumenfeld, A.; Meyer, S.; Namburi, P.; Morad, Y.; Gradstein, L.; Swaroop, A.; Banin, E.; Sharon, D. Genetic Analysis of the Rhodopsin Gene Identifies a Mosaic Dominant Retinitis Pigmentosa Mutation in a Healthy Individual. Investig. Ophthalmol. Vis. Sci. 2016, 57, 940–947.
- Xiao, T.; Xu, K.; Zhang, X.; Xie, Y.; Li, Y. Sector Retinitis Pigmentosa caused by mutations of the RHO gene. Eye 2019, 33, 592–599.
- Dryja, T.P.; Mukai, S.; Petersen, R.; Rapaport, J.M.; Walton, D.; Yandell, D.W. Parental origin of mutations of the retinoblastoma gene. Nature 1989, 339, 556–558.
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Hayes, K.C.; Nicholson, B.W.; Weigel-DiFranco, C.; Willett, W. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch. Ophthalmol. 1993, 111, 761–772.
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Weigel-DiFranco, C.; Willett, W.C. ω-3 intake and visual acuity in patients with retinitis pigmentosa receiving vitamin A. Arch. Ophthalmol. 2012, 130, 707–711.
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Weigel-DiFranco, C.; Brockhurst, R.J.; Hayes, K.C.; Johnson, E.J.; Anderson, E.J.; Johnson, C.A.; Gaudio, A.R.; et al. Clinical trial of lutein in patients with retinitis pigmentosa receiving vitamin A. Arch. Ophthalmol. 2010, 128, 403–411.
- Bahrami, H.; Melia, M.; Dagnelie, G. Lutein supplementation in retinitis pigmentosa: PC-based vision assessment in a randomized double-masked placebo-controlled clinical trial . BMC Ophthalmol. 2006, 6, 23.
- Parker, R.O.; Crouch, R.K. Retinol dehydrogenases (RDHs) in the visual cycle. Exp. Eye Res. 2010, 91, 788–792.
- Simon, A.; Lagercrantz, J.; Bajalica-Lagercrantz, S.; Eriksson, U. Primary structure of human 11-cis retinol dehydrogenase and organization and chromosomal localization of the corresponding gene. Genomics 1996, 36, 424–430.
- Simon, A.; Hellman, U.; Wernstedt, C.; Eriksson, U. The retinal pigment epithelial-specific 11-cis retinol dehydrogenase belongs to the family of short chain alcohol dehydrogenases. J. Biol. Chem. 1995, 270, 1107–1112.
- Krill, A.E.; Klien, B.A. Flecked Retina Syndrome. Arch. Ophthalmol. 1965, 74, 496–508.
- Yamamoto, H.; Simon, A.; Eriksson, U.; Harris, E.; Berson, E.L.; Dryja, T.P. Mutations in the gene encoding 11-cis retinol dehydrogenase cause delayed dark adaptation and fundus albipunctatus. Nat. Genet. 1999, 22, 188–191.
- Sergouniotis, P.I.; Sohn, E.H.; Li, Z.; McBain, V.A.; Wright, G.A.; Moore, A.T.; Robson, A.G.; Holder, G.E.; Webster, A.R. Phenotypic variability in RDH5 retinopathy (Fundus Albipunctatus). Ophthalmology 2011, 118, 1661–1670.
- Katagiri, S.; Hayashi, T.; Nakamura, M.; Mizobuchi, K.; Gekka, T.; Komori, S.; Ueno, S.; Terasaki, H.; Sakuramoto, H.; Kuniyoshi, K.; et al. RDH5-Related Fundus Albipunctatus in a Large Japanese Cohort. Investig. Ophthalmol. Vis. Sci. 2020, 61, 53.
- Schatz, P.; Preising, M.; Lorenz, B.; Sander, B.; Larsen, M.; Eckstein, C.; Rosenberg, T. Lack of autofluorescence in fundus albipunctatus associated with mutations in RDH5. Retina 2010, 30, 1704–1713.
- Skorczyk-Werner, A.; Pawłowski, P.; Michalczuk, M.; Warowicka, A.; Wawrocka, A.; Wicher, K.; Bakunowicz-Łazarczyk, A.; Krawczyński, M.R. Fundus albipunctatus: Review of the literature and report of a novel RDH5 gene mutation affecting the invariant tyrosine (p.Tyr175Phe). J. Appl. Genet. 2015, 56, 317–327.
- Driessen, C.A.; Winkens, H.J.; Hoffmann, K.; Kuhlmann, L.D.; Janssen, B.P.; Van Vugt, A.H.; Van Hooser, J.P.; Wieringa, B.E.; Deutman, A.F.; Palczewski, K.; et al. Disruption of the 11-cis-retinol dehydrogenase gene leads to accumulation of cis-retinols and cis-retinyl esters. Mol. Cell Biol. 2000, 20, 4275–4287.
- Niwa, Y.; Kondo, M.; Ueno, S.; Nakamura, M.; Terasaki, H.; Miyake, Y. Cone and Rod Dysfunction in Fundus Albipunctatus with RDH5 Mutation: An Electrophysiological Study. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1480–1485.
- Rotenstreich, Y.; Harats, D.; Shaish, A.; Pras, E.; Belkin, M. Treatment of a retinal dystrophy, fundus albipunctatus, with oral 9-cis-β-carotene. Br. J. Ophthalmol. 2010, 94, 616–621.
- Sahu, B.; Maeda, A. Retinol Dehydrogenases Regulate Vitamin A Metabolism for Visual Function. Nutrients 2016, 8, 746.
- Haeseleer, F.; Jang, G.-F.; Imanishi, Y.; Driessen, C.A.G.G.; Matsumura, M.; Nelson, P.S.; Palczewski, K. Dual-substrate specificity short chain retinol dehydrogenases from the vertebrate retina. J. Biol. Chem. 2002, 277, 45537–45546.
- Kim, T.S.; Maeda, A.; Maeda, T.; Heinlein, C.; Kedishvili, N.; Palczewski, K.; Nelson, P.S. Delayed dark adaptation in 11-cis-retinol dehydrogenase-deficient mice: A role of RDH11 in visual processes in vivo. J. Biol. Chem. 2005, 280, 8694–8704.
- Xie, Y.A.; Lee, W.; Cai, C.; Gambin, T.; Nõupuu, K.; Sujirakul, T.; Ayuso, C.; Jhangiani, S.; Muzny, D.; Boerwinkle, E.; et al. New syndrome with retinitis pigmentosa is caused by nonsense mutations in retinol dehydrogenase RDH11. Hum. Mol. Genet. 2014, 23, 5774–5780.
- Kiser, P.D.; Golczak, M.; Maeda, A.; Palczewski, K. Key enzymes of the retinoid (visual) cycle in vertebrate retina. Biochim. Biophys. Acta 2012, 1821, 137–151.
- Saari, J.C. Vitamin A metabolism in rod and cone visual cycles. Annu. Rev. Nutr. 2012, 32, 125–145.
- Garg, A.; Lee, W.; Sengillo, J.D.; Allikmets, R.; Garg, K.; Tsang, S.H. Peripapillary sparing in RDH12-associated Leber congenital amaurosis. Ophthalmic Genet. 2017, 38, 575–579.
- Daruwalla, A.; Choi, E.H.; Palczewski, K.; Kiser, P.D. Structural biology of 11-cis-retinaldehyde production in the classical visual cycle. Biochem. J. 2018, 475, 3171–3188.
- Ba-Abbad, R.; Arno, G.; Robson, A.G.; Bouras, K.; Georgiou, M.; Wright, G.; Webster, A.R.; Michaelides, M. Macula-predominant retinopathy associated with biallelic variants in RDH12. Ophthalmic Genet. 2020, 41, 612–615.
- Mackay, D.S.; Dev Borman, A.; Moradi, P.; Henderson, R.H.; Li, Z.; Wright, G.A.; Waseem, N.; Gandra, M.; Thompson, D.A.; Bhattacharya, S.S.; et al. RDH12 retinopathy: Novel mutations and phenotypic description. Mol. Vis. 2011, 17, 2706–2716.
- Fahim, A.T.; Bouzia, Z.; Branham, K.H.; Kumaran, N.; Vargas, M.E.; Feathers, K.L.; Perera, N.D.; Young, K.; Khan, N.W.; Heckenlively, J.R.; et al. Detailed clinical characterisation, unique features and natural history of autosomal recessive RDH12 associated retinal degeneration. Br. J. Ophthalmol. 2019, 103, 1789.
- Scott, H.A.; Place, E.M.; Ferenchak, K.; Zampaglione, E.; Wagner, N.E.; Chao, K.R.; DiTroia, S.P.; Navarro-Gomez, D.; Mukai, S.; Huckfeldt, R.M.; et al. Expanding the phenotypic spectrum in RDH12-associated retinal disease. Cold Spring Harb. Mol. Case Stud. 2020, 6, a004754.
- Bunt-Milam, A.H.; Saari, J.C. Immunocytochemical localization of two retinoid-binding proteins in vertebrate retina. J. Cell Biol. 1983, 97, 703–712.
- Bernal, S.; Calaf, M.; Adan, A.; Solans, T.; Valverde, D.; Ayuso, C.; Baiget, M. Evaluation of RLBP1 in 50 autosomal recessive retinitis pigmentosa and 4 retinitis punctata albescens Spanish families. Ophthalmic Genet. 2001, 22, 19–25.
- Stecher, H.; Gelb, M.H.; Saari, J.C.; Palczewski, K. Preferential release of 11-cis-retinol from retinal pigment epithelial cells in the presence of cellular retinaldehyde-binding protein. J. Biol. Chem. 1999, 274, 8577–8585.
- Winston, A.; Rando, R.R. Regulation of isomerohydrolase activity in the visual cycle. Biochemistry 1998, 37, 2044–2050.
- Kolesnikov, A.V.; Kiser, P.D.; Palczewski, K.; Kefalov, V.J. Function of mammalian M-cones depends on the level of CRALBP in Müller cells. J. Gen. Physiol. 2021, 153, e202012675.
- Xue, Y.; Shen, S.Q.; Jui, J.; Rupp, A.C.; Byrne, L.C.; Hattar, S.; Flannery, J.G.; Corbo, J.C.; Kefalov, V.J. CRALBP supports the mammalian retinal visual cycle and cone vision. J. Clin. Investig. 2015, 125, 727–738.
- Lima de Carvalho, J.R., Jr.; Kim, H.J.; Ueda, K.; Zhao, J.; Owji, A.P.; Yang, T.; Tsang, S.H.; Sparrow, J.R. Effects of deficiency in the RLBP1-encoded visual cycle protein CRALBP on visual dysfunction in humans and mice. J. Biol. Chem. 2020, 295, 6767–6780.
- Torres-Costa, S.; Ferreira, C.S.; Grangeia, A.; Santos-Silva, R.; Brandão, E.; Estrela-Silva, S.; Falcão-Reis, F. A novel homozygous frameshift variant in the cellular retinaldehyde-binding protein 1 (RLBP1) gene causes retinitis punctata albescens. Eur. J. Ophthalmol. 2021, 31, NP74–NP80.
- Scimone, C.; Donato, L.; Esposito, T.; Rinaldi, C.; D’Angelo, R.; Sidoti, A. A novel RLBP1 gene geographical area-related mutation present in a young patient with retinitis punctata albescens. Hum. Genom. 2017, 11, 18.
- Dessalces, E.; Bocquet, B.; Bourien, J.; Zanlonghi, X.; Verdet, R.; Meunier, I.; Hamel, C.P. Early-Onset Foveal Involvement in Retinitis Punctata Albescens With Mutations in RLBP1. JAMA Ophthalmol. 2013, 131, 1314–1323.
- Bocquet, B.; El Alami Trebki, H.; Roux, A.F.; Labesse, G.; Brabet, P.; Arndt, C.; Zanlonghi, X.; Defoort-Dhellemmes, S.; Hamroun, D.; Boulicot-Séguin, C.; et al. Retinitis Punctata Albescens and RLBP1-Allied Phenotypes: Phenotype–Genotype Correlation and Natural History in the Aim of Gene Therapy. Ophthalmol. Sci. 2021, 1, 100052.
- Farrar, G.J.; Carrigan, M.; Dockery, A.; Millington-Ward, S.; Palfi, A.; Chadderton, N.; Humphries, M.; Kiang, A.S.; Kenna, P.F.; Humphries, P. Toward an elucidation of the molecular genetics of inherited retinal degenerations. Hum. Mol. Genet. 2017, 26, R2–R11.
- Kiser, P.D.; Palczewski, K. Retinoids and Retinal Diseases. Annu. Rev. Vis. Sci. 2016, 2, 197–234.
- Perusek, L.; Maeda, T. Vitamin A derivatives as treatment options for retinal degenerative diseases. Nutrients 2013, 5, 2646–2666.
- Zeng, S.; Zhang, T.; Madigan, M.C.; Fernando, N.; Aggio-Bruce, R.; Zhou, F.; Pierce, M.; Chen, Y.; Huang, L.; Natoli, R.; et al. Interphotoreceptor Retinoid-Binding Protein (IRBP) in Retinal Health and Disease. Front. Cell. Neurosci. 2020, 14, 577935.
- Parker, R.O.; Crouch, R.K. The interphotoreceptor retinoid binding (IRBP) is essential for normal retinoid processing in cone photoreceptors. Adv. Exp. Med. Biol. 2010, 664, 141–149.
- Li, S.; Yang, Z.; Hu, J.; Gordon, W.C.; Bazan, N.G.; Haas, A.L.; Bok, D.; Jin, M. Secretory Defect and Cytotoxicity: The Potential Disease Mechanisms for the Retinitis Pigmentosa (RP)-Associated Interphotoreceptor Retinoid-Binding Protein (IRBP). J. Biol. Chem. 2013, 288, 11395–11406.
- den Hollander, A.I.; McGee, T.L.; Ziviello, C.; Banfi, S.; Dryja, T.P.; Gonzalez-Fernandez, F.; Ghosh, D.; Berson, E.L. A homozygous missense mutation in the IRBP gene (RBP3) associated with autosomal recessive retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1864–1872.
- Arno, G.; Hull, S.; Robson, A.G.; Holder, G.E.; Cheetham, M.E.; Webster, A.R.; Plagnol, V.; Moore, A.T. Lack of Interphotoreceptor Retinoid Binding Protein Caused by Homozygous Mutation of RBP3 Is Associated with High Myopia and Retinal Dystrophy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2358–2365.
- van Bennekum, A.M.; Wei, S.; Gamble, M.V.; Vogel, S.; Piantedosi, R.; Gottesman, M.; Episkopou, V.; Blaner, W.S. Biochemical basis for depressed serum retinol levels in transthyretin-deficient mice. J. Biol. Chem. 2001, 276, 1107–1113.
- Berry, D.C.; Croniger, C.M.; Ghyselinck, N.B.; Noy, N. Transthyretin blocks retinol uptake and cell signaling by the holo-retinol-binding protein receptor STRA6. Mol. Cell. Biol. 2012, 32, 3851–3859.
- Seeliger, M.W.; Biesalski, H.K.; Wissinger, B.; Gollnick, H.; Gielen, S.; Frank, J.; Beck, S.; Zrenner, E. Phenotype in retinol deficiency due to a hereditary defect in retinol binding protein synthesis. Investig. Ophthalmol. Vis. Sci. 1999, 40, 3–11.
- Biesalski, H.K.; Frank, J.; Beck, S.C.; Heinrich, F.; Illek, B.; Reifen, R.; Gollnick, H.; Seeliger, M.W.; Wissinger, B.; Zrenner, E. Biochemical but not clinical vitamin A deficiency results from mutations in the gene for retinol binding protein. Am. J. Clin. Nutr. 1999, 69, 931–936.
- Cukras, C.; Gaasterland, T.; Lee, P.; Gudiseva, H.V.; Chavali, V.R.; Pullakhandam, R.; Maranhao, B.; Edsall, L.; Soares, S.; Reddy, G.B.; et al. Exome analysis identified a novel mutation in the RBP4 gene in a consanguineous pedigree with retinal dystrophy and developmental abnormalities. PLoS ONE 2012, 7, e50205.
- Khan, K.N.; Carss, K.; Raymond, F.L.; Islam, F.; Nihr BioResource-Rare Diseases, C.; Moore, A.T.; Michaelides, M.; Arno, G. Vitamin A deficiency due to bi-allelic mutation of RBP4: There’s more to it than meets the eye. Ophthalmic Genet. 2017, 38, 465–466.
- Chou, C.M.; Nelson, C.; Tarlé, S.A.; Pribila, J.T.; Bardakjian, T.; Woods, S.; Schneider, A.; Glaser, T. Biochemical Basis for Dominant Inheritance, Variable Penetrance, and Maternal Effects in RBP4 Congenital Eye Disease. Cell 2015, 161, 634–646.
- Cehajic-Kapetanovic, J.; Jasani, K.M.; Shanks, M.; Clouston, P.; MacLaren, R.E. A novel homozygous c.67C>T variant in retinol binding protein 4 (RBP4) associated with retinitis pigmentosa and childhood acne vulgaris. Ophthalmic Genet. 2020, 41, 288–292.
- Chen, X.N.; Korenberg, J.R.; Jiang, M.; Shen, D.; Fong, H.K. Localization of the human RGR opsin gene to chromosome 10q23. Hum. Genet. 1996, 97, 720–722.
- Jiang, M.; Pandey, S.; Fong, H.K. An opsin homologue in the retina and pigment epithelium. Investig. Ophthalmol. Vis. Sci. 1993, 34, 3669–3678.
- Pandey, S.; Blanks, J.C.; Spee, C.; Jiang, M.; Fong, H.K. Cytoplasmic retinal localization of an evolutionary homolog of the visual pigments. Exp. Eye Res. 1994, 58, 605–613.
- Li, J.; Xiao, X.; Li, S.; Jia, X.; Guo, X.; Zhang, Q. RGR variants in different forms of retinal diseases: The undetermined role of truncation mutations. Mol. Med. Rep. 2016, 14, 4811–4815.
- Morimura, H.; Saindelle-Ribeaudeau, F.; Berson, E.L.; Dryja, T.P. Mutations in RGR, encoding a light-sensitive opsin homologue, in patients with retinitis pigmentosa. Nat. Genet. 1999, 23, 393–394.
- Ba-Abbad, R.; Leys, M.; Wang, X.; Chakarova, C.; Waseem, N.; Carss, K.J.; Raymond, F.L.; Bujakowska, K.M.; Pierce, E.A.; Mahroo, O.A.; et al. Clinical Features of a Retinopathy Associated With a Dominant Allele of the RGR Gene. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4812–4820.
- Huang, C.-H.; Yang, C.-M.; Yang, C.-H.; Hou, Y.-C.; Chen, T.-C. Leber’s Congenital Amaurosis: Current Concepts of Genotype-Phenotype Correlations. Genes 2021, 12, 1261.
- Sears, A.E.; Palczewski, K. Lecithin:Retinol Acyltransferase: A Key Enzyme Involved in the Retinoid (visual) Cycle. Biochemistry 2016, 55, 3082–3091.
- Chen, Y.; Huang, L.; Jiao, X.; Riazuddin, S.; Riazuddin, S.A.; Fielding Hetmancik, J. A novel LRAT mutation affecting splicing in a family with early onset retinitis pigmentosa. Hum. Genom. 2018, 12, 35.
- Dev Borman, A.; Ocaka, L.A.; Mackay, D.S.; Ripamonti, C.; Henderson, R.H.; Moradi, P.; Hall, G.; Black, G.C.; Robson, A.G.; Holder, G.E.; et al. Early onset retinal dystrophy due to mutations in LRAT: Molecular analysis and detailed phenotypic study. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3927–3938.
- Saini, A.; Almasarweh, S.; Acosta, S.; Jayakar, P.; Janvier, M.; Wong, T.C.; Salyakina, D.; Sasaki, J. Syndromic Microphthalmia 9: Role of rapid genome sequencing and novel mutations in STRA6 gene. Prog. Pediatric Cardiol. 2021, 101443.
- Ruiz, A.; Mark, M.; Jacobs, H.; Klopfenstein, M.; Hu, J.; Lloyd, M.; Habib, S.; Tosha, C.; Radu, R.A.; Ghyselinck, N.B.; et al. Retinoid Content, Visual Responses, and Ocular Morphology Are Compromised in the Retinas of Mice Lacking the Retinol-Binding Protein Receptor, STRA6. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3027–3039.
- Kawaguchi, R.; Zhong, M.; Kassai, M.; Ter-Stepanian, M.; Sun, H. STRA6-catalyzed vitamin A influx, efflux, and exchange. J. Membr. Biol. 2012, 245, 731–745.
- Chassaing, N.; Golzio, C.; Odent, S.; Lequeux, L.; Vigouroux, A.; Martinovic-Bouriel, J.; Tiziano, F.D.; Masini, L.; Piro, F.; Maragliano, G.; et al. Phenotypic spectrum of STRA6 mutations: From Matthew-Wood syndrome to non-lethal anophthalmia. Hum. Mutat. 2009, 30, E673–E681.
- Pasutto, F.; Flinter, F.; Rauch, A.; Reis, A. Novel STRA6 null mutations in the original family described with Matthew-Wood syndrome. Am. J. Med. Genet. A 2018, 176, 134–138.
- Pasutto, F.; Sticht, H.; Hammersen, G.; Gillessen-Kaesbach, G.; Fitzpatrick, D.R.; Nürnberg, G.; Brasch, F.; Schirmer-Zimmermann, H.; Tolmie, J.L.; Chitayat, D.; et al. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am. J. Hum. Genet. 2007, 80, 550–560.
- Golzio, C.; Martinovic-Bouriel, J.; Thomas, S.; Mougou-Zrelli, S.; Grattagliano-Bessieres, B.; Bonniere, M.; Delahaye, S.; Munnich, A.; Encha-Razavi, F.; Lyonnet, S.; et al. Matthew-Wood syndrome is caused by truncating mutations in the retinol-binding protein receptor gene STRA6. Am. J. Hum. Genet. 2007, 80, 1179–1187.
- Christensen, D.R.G.; Brown, F.E.; Cree, A.J.; Ratnayaka, J.A.; Lotery, A.J. Sorsby fundus dystrophy—A review of pathology and disease mechanisms. Exp. Eye Res. 2017, 165, 35–46.
- Weber, B.H.; Vogt, G.; Pruett, R.C.; Stöhr, H.; Felbor, U. Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with Sorsby’s fundus dystrophy. Nat. Genet. 1994, 8, 352–356.
- Gliem, M.; Müller, P.L.; Mangold, E.; Holz, F.G.; Bolz, H.J.; Stöhr, H.; Weber, B.H.; Charbel Issa, P. Sorsby Fundus Dystrophy: Novel Mutations, Novel Phenotypic Characteristics, and Treatment Outcomes. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2664–2676.
- Sorsby, A.; Mason, M.E. A fundus dystrophy with unusual features. Br. J. Ophthalmol. 1949, 33, 67–97.
- Baston, A.; Gerhardt, C.; Zandi, S.; Garweg, J.G. Visual Outcome after Intravitreal Anti-VEGF Therapy for Macular Neovascularisation Secondary to Sorsby’s Fundus Dystrophy: A Systematic Review. J. Clin. Med. 2021, 10, 2433.
- Naessens, S.; De Zaeytijd, J.; Syx, D.; Vandenbroucke, R.E.; Smeets, F.; Van Cauwenbergh, C.; Leroy, B.P.; Peelman, F.; Coppieters, F. The N-terminal p.(Ser38Cys) TIMP3 mutation underlying Sorsby fundus dystrophy is a founder mutation disrupting an intramolecular disulfide bond. Hum. Mutat. 2019, 40, 539–551.
- Gourier, H.C.Y.; Chong, N.V. Can Novel Treatment of Age-Related Macular Degeneration Be Developed by Better Understanding of Sorsby’s Fundus Dystrophy. J. Clin. Med. 2015, 4, 874–883.
- Chang, A.A.; Zhu, M.; Billson, F. The interaction of indocyanine green with human retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1463–1467.